Here, we explain how to visualize VeloxChem results in Jupyter notebooks.
The underlying calculation can be obtained either directly in a notebook or imported from a VeloxChem .h5 file.
import veloxchem as vlxMolecular structure¶
In Jupyter notebook, a molecule object can be visualized using the show function.
The atom_indices and atom_labels options can be used.
From a notebook cell
molecule = vlx.Molecule.read_smiles('C1=CC=C(C=C1)C(=O)O')
molecule.show(atom_indices=True, atom_labels=True)Loading...
From an h5 file
biph_dict = vlx.read_results("../output_files/biphenyl-scf.h5", label="scf")
biph = vlx.Molecule.from_dict(biph_dict)
biph.show()Loading...
Structure optimization¶
From notebook cell
molecule = vlx.Molecule.read_smiles('CCO')
basis = vlx.MolecularBasis.read(molecule, 'def2-svp')
scf_drv = vlx.ScfRestrictedDriver()
scf_drv.xcfun = 'b3lyp'
results = scf_drv.compute(molecule, basis)
opt_drv = vlx.OptimizationDriver(scf_drv)
opt_results = opt_drv.compute(molecule, basis, results)
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Kohn-Sham
Initial Guess Model : Superposition of Atomic Densities
Convergence Accelerator : Two Level Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Nuclear repulsion energy: 81.7275742680 a.u.
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Molecular grid with 115446 points generated in 0.04 sec.
* Info * Overlap matrix computed in 0.00 sec.
* Info * Kinetic energy matrix computed in 0.00 sec.
* Info * Nuclear potential matrix computed in 0.00 sec.
* Info * Orthogonalization matrix computed in 0.00 sec.
* Info * Starting Reduced Basis SCF calculation...
* Info * ...done. SCF energy in reduced basis set: -153.878486434045 a.u. Time: 0.07 sec.
* Info * Overlap matrix computed in 0.00 sec.
* Info * Kinetic energy matrix computed in 0.00 sec.
* Info * Nuclear potential matrix computed in 0.00 sec.
* Info * Orthogonalization matrix computed in 0.00 sec.
Iter. | Kohn-Sham Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -154.914063112834 0.0000000000 0.25703983 0.02179405 0.00000000
2 -154.916286182150 -0.0022230693 0.20417296 0.01300491 0.15244838
3 -154.921403293175 -0.0051171110 0.03363328 0.00174044 0.07260922
4 -154.921522515308 -0.0001192221 0.01000619 0.00081150 0.01114472
5 -154.921531424625 -0.0000089093 0.00210424 0.00013483 0.00355082
6 -154.921531845218 -0.0000004206 0.00034752 0.00002790 0.00073932
7 -154.921531860515 -0.0000000153 0.00004173 0.00000246 0.00015462
8 -154.921531860689 -0.0000000002 0.00001994 0.00000104 0.00001747
9 -154.921531860738 -0.0000000000 0.00000195 0.00000010 0.00000661
10 -154.921531860738 -0.0000000000 0.00000016 0.00000001 0.00000088
*** SCF converged in 10 iterations. Time: 1.12 sec.
Spin-Restricted Kohn-Sham:
--------------------------
Total Energy : -154.9215318607 a.u.
Electronic Energy : -236.6491061287 a.u.
Nuclear Repulsion Energy : 81.7275742680 a.u.
------------------------------------
Gradient Norm : 0.0000001635 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
Spin Restricted Orbitals
------------------------
Molecular Orbital No. 9:
--------------------------
Occupation: 2.000 Energy: -0.42032 a.u.
( 1 C 1p0 : -0.34) ( 1 C 2p0 : -0.15) ( 2 C 1p-1: -0.22)
( 2 C 1p0 : 0.22) ( 3 O 1p+1: -0.24) ( 6 H 1s : -0.21)
Molecular Orbital No. 10:
--------------------------
Occupation: 2.000 Energy: -0.38156 a.u.
( 1 C 1p-1: -0.38) ( 1 C 2p-1: -0.17) ( 3 O 1p-1: 0.21)
( 4 H 1s : 0.33) ( 6 H 1s : -0.17)
Molecular Orbital No. 11:
--------------------------
Occupation: 2.000 Energy: -0.36887 a.u.
( 1 C 1p+1: 0.33) ( 2 C 1p+1: -0.21) ( 3 O 1p-1: 0.16)
( 3 O 1p0 : 0.24) ( 5 H 1s : -0.30) ( 6 H 1s : 0.22)
( 7 H 1s : -0.18)
Molecular Orbital No. 12:
--------------------------
Occupation: 2.000 Energy: -0.31907 a.u.
( 2 C 1p-1: 0.21) ( 2 C 1p0 : 0.17) ( 3 O 3s : 0.21)
( 3 O 1p-1: -0.26) ( 3 O 1p0 : -0.27) ( 3 O 2p-1: -0.17)
( 3 O 2p0 : -0.19) ( 7 H 1s : -0.25) ( 7 H 2s : -0.16)
( 8 H 1s : 0.25) ( 8 H 2s : 0.16) ( 9 H 1s : -0.19)
Molecular Orbital No. 13:
--------------------------
Occupation: 2.000 Energy: -0.26551 a.u.
( 1 C 3s : -0.19) ( 1 C 1p0 : 0.21) ( 3 O 1p+1: -0.23)
( 3 O 1p-1: -0.43) ( 3 O 1p0 : 0.31) ( 3 O 2p+1: -0.16)
( 3 O 2p-1: -0.32) ( 3 O 2p0 : 0.25) ( 8 H 1s : 0.16)
Molecular Orbital No. 14:
--------------------------
Occupation: 0.000 Energy: 0.04208 a.u.
( 1 C 3s : 0.93) ( 1 C 2p+1: -0.26) ( 1 C 2p0 : 0.20)
( 2 C 3s : 0.93) ( 2 C 2p-1: 0.36) ( 3 O 3s : 0.56)
( 3 O 1p+1: -0.19) ( 3 O 2p+1: -0.28) ( 4 H 2s : -0.35)
( 5 H 2s : -0.66) ( 6 H 2s : -0.43) ( 7 H 2s : -0.31)
( 8 H 2s : -0.80) ( 9 H 2s : -0.93)
Molecular Orbital No. 15:
--------------------------
Occupation: 0.000 Energy: 0.08398 a.u.
( 1 C 3s : -1.82) ( 1 C 2p-1: 0.17) ( 1 C 2p0 : -0.28)
( 2 C 3s : -0.63) ( 2 C 2p+1: -0.43) ( 2 C 2p0 : 0.30)
( 3 O 3s : 0.52) ( 3 O 2p-1: 0.16) ( 4 H 2s : 0.93)
( 5 H 2s : 0.48) ( 6 H 2s : 1.03) ( 7 H 2s : 1.01)
( 8 H 2s : 0.18) ( 9 H 2s : -0.74)
Molecular Orbital No. 16:
--------------------------
Occupation: 0.000 Energy: 0.10656 a.u.
( 1 C 3s : 2.24) ( 1 C 2p+1: -0.29) ( 1 C 2p0 : -0.28)
( 2 C 3s : -2.18) ( 2 C 2p+1: -0.31) ( 2 C 2p0 : -0.41)
( 4 H 2s : -0.85) ( 5 H 2s : -1.32) ( 6 H 2s : -0.17)
( 7 H 2s : 1.22) ( 8 H 2s : 1.10)
Molecular Orbital No. 17:
--------------------------
Occupation: 0.000 Energy: 0.12092 a.u.
( 1 C 1p-1: 0.19) ( 1 C 2p+1: -0.22) ( 1 C 2p-1: 0.52)
( 1 C 2p0 : 0.26) ( 2 C 3s : 0.48) ( 2 C 1p+1: -0.18)
( 2 C 1p-1: 0.22) ( 2 C 2p+1: -0.52) ( 2 C 2p-1: 0.57)
( 3 O 3s : -0.48) ( 3 O 2p-1: -0.19) ( 4 H 2s : 0.98)
( 5 H 2s : -0.83) ( 6 H 2s : -0.31) ( 7 H 2s : 0.83)
( 8 H 2s : -1.35) ( 9 H 2s : 0.51)
Molecular Orbital No. 18:
--------------------------
Occupation: 0.000 Energy: 0.13483 a.u.
( 1 C 3s : -0.53) ( 1 C 1p+1: -0.23) ( 1 C 1p0 : -0.22)
( 1 C 2p+1: -0.85) ( 1 C 2p-1: -0.51) ( 1 C 2p0 : -0.67)
( 2 C 3s : 0.95) ( 2 C 2p+1: 0.30) ( 2 C 2p-1: 0.23)
( 4 H 2s : -0.61) ( 5 H 2s : -0.66) ( 6 H 2s : 1.94)
( 7 H 2s : -0.59) ( 8 H 2s : -0.64) ( 9 H 2s : 0.21)
Ground State Dipole Moment
----------------------------
X : -0.242040 a.u. -0.615205 Debye
Y : 0.403846 a.u. 1.026473 Debye
Z : 0.395932 a.u. 1.006358 Debye
Total : 0.615172 a.u. 1.563611 Debye
Optimization Driver Setup
===========================
Coordinate System : TRIC
Constraints : No
Max. Number of Steps : 300
Transition State : No
IRC : No
Hessian : never
* Info * Using geomeTRIC for geometry optimization.
L.-P. Wang and C.C. Song, J. Chem. Phys. 2016, 144, 214108
* Info * Computing energy and gradient...
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Kohn-Sham
Initial Guess Model : Superposition of Atomic Densities
Convergence Accelerator : Two Level Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Nuclear repulsion energy: 81.7275742680 a.u.
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Molecular grid with 115446 points generated in 0.04 sec.
* Info * Overlap matrix computed in 0.00 sec.
* Info * Kinetic energy matrix computed in 0.00 sec.
* Info * Nuclear potential matrix computed in 0.00 sec.
* Info * Orthogonalization matrix computed in 0.00 sec.
* Info * Starting Reduced Basis SCF calculation...
* Info * ...done. SCF energy in reduced basis set: -153.878486434045 a.u. Time: 0.07 sec.
* Info * Overlap matrix computed in 0.00 sec.
* Info * Kinetic energy matrix computed in 0.00 sec.
* Info * Nuclear potential matrix computed in 0.00 sec.
* Info * Orthogonalization matrix computed in 0.00 sec.
Iter. | Kohn-Sham Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -154.914063112835 0.0000000000 0.25703983 0.02179405 0.00000000
2 -154.916286182150 -0.0022230693 0.20417296 0.01300491 0.15244838
3 -154.921403293175 -0.0051171110 0.03363328 0.00174044 0.07260922
4 -154.921522515308 -0.0001192221 0.01000619 0.00081150 0.01114472
5 -154.921531424624 -0.0000089093 0.00210424 0.00013483 0.00355082
6 -154.921531845218 -0.0000004206 0.00034752 0.00002790 0.00073932
7 -154.921531860515 -0.0000000153 0.00004173 0.00000246 0.00015462
8 -154.921531860689 -0.0000000002 0.00001994 0.00000104 0.00001747
9 -154.921531860738 -0.0000000000 0.00000195 0.00000010 0.00000661
10 -154.921531860738 -0.0000000000 0.00000016 0.00000001 0.00000088
* Info * Checkpoint written to file: vlx_20251015_6d4463a8_scf.h5
*** SCF converged in 10 iterations. Time: 1.16 sec.
Spin-Restricted Kohn-Sham:
--------------------------
Total Energy : -154.9215318607 a.u.
Electronic Energy : -236.6491061287 a.u.
Nuclear Repulsion Energy : 81.7275742680 a.u.
------------------------------------
Gradient Norm : 0.0000001635 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
Spin Restricted Orbitals
------------------------
Molecular Orbital No. 9:
--------------------------
Occupation: 2.000 Energy: -0.42032 a.u.
( 1 C 1p0 : -0.34) ( 1 C 2p0 : -0.15) ( 2 C 1p-1: -0.22)
( 2 C 1p0 : 0.22) ( 3 O 1p+1: -0.24) ( 6 H 1s : -0.21)
Molecular Orbital No. 10:
--------------------------
Occupation: 2.000 Energy: -0.38156 a.u.
( 1 C 1p-1: -0.38) ( 1 C 2p-1: -0.17) ( 3 O 1p-1: 0.21)
( 4 H 1s : 0.33) ( 6 H 1s : -0.17)
Molecular Orbital No. 11:
--------------------------
Occupation: 2.000 Energy: -0.36887 a.u.
( 1 C 1p+1: 0.33) ( 2 C 1p+1: -0.21) ( 3 O 1p-1: 0.16)
( 3 O 1p0 : 0.24) ( 5 H 1s : -0.30) ( 6 H 1s : 0.22)
( 7 H 1s : -0.18)
Molecular Orbital No. 12:
--------------------------
Occupation: 2.000 Energy: -0.31907 a.u.
( 2 C 1p-1: 0.21) ( 2 C 1p0 : 0.17) ( 3 O 3s : 0.21)
( 3 O 1p-1: -0.26) ( 3 O 1p0 : -0.27) ( 3 O 2p-1: -0.17)
( 3 O 2p0 : -0.19) ( 7 H 1s : -0.25) ( 7 H 2s : -0.16)
( 8 H 1s : 0.25) ( 8 H 2s : 0.16) ( 9 H 1s : -0.19)
Molecular Orbital No. 13:
--------------------------
Occupation: 2.000 Energy: -0.26551 a.u.
( 1 C 3s : -0.19) ( 1 C 1p0 : 0.21) ( 3 O 1p+1: -0.23)
( 3 O 1p-1: -0.43) ( 3 O 1p0 : 0.31) ( 3 O 2p+1: -0.16)
( 3 O 2p-1: -0.32) ( 3 O 2p0 : 0.25) ( 8 H 1s : 0.16)
Molecular Orbital No. 14:
--------------------------
Occupation: 0.000 Energy: 0.04208 a.u.
( 1 C 3s : 0.93) ( 1 C 2p+1: -0.26) ( 1 C 2p0 : 0.20)
( 2 C 3s : 0.93) ( 2 C 2p-1: 0.36) ( 3 O 3s : 0.56)
( 3 O 1p+1: -0.19) ( 3 O 2p+1: -0.28) ( 4 H 2s : -0.35)
( 5 H 2s : -0.66) ( 6 H 2s : -0.43) ( 7 H 2s : -0.31)
( 8 H 2s : -0.80) ( 9 H 2s : -0.93)
Molecular Orbital No. 15:
--------------------------
Occupation: 0.000 Energy: 0.08398 a.u.
( 1 C 3s : -1.82) ( 1 C 2p-1: 0.17) ( 1 C 2p0 : -0.28)
( 2 C 3s : -0.63) ( 2 C 2p+1: -0.43) ( 2 C 2p0 : 0.30)
( 3 O 3s : 0.52) ( 3 O 2p-1: 0.16) ( 4 H 2s : 0.93)
( 5 H 2s : 0.48) ( 6 H 2s : 1.03) ( 7 H 2s : 1.01)
( 8 H 2s : 0.18) ( 9 H 2s : -0.74)
Molecular Orbital No. 16:
--------------------------
Occupation: 0.000 Energy: 0.10656 a.u.
( 1 C 3s : 2.24) ( 1 C 2p+1: -0.29) ( 1 C 2p0 : -0.28)
( 2 C 3s : -2.18) ( 2 C 2p+1: -0.31) ( 2 C 2p0 : -0.41)
( 4 H 2s : -0.85) ( 5 H 2s : -1.32) ( 6 H 2s : -0.17)
( 7 H 2s : 1.22) ( 8 H 2s : 1.10)
Molecular Orbital No. 17:
--------------------------
Occupation: 0.000 Energy: 0.12092 a.u.
( 1 C 1p-1: 0.19) ( 1 C 2p+1: -0.22) ( 1 C 2p-1: 0.52)
( 1 C 2p0 : 0.26) ( 2 C 3s : 0.48) ( 2 C 1p+1: -0.18)
( 2 C 1p-1: 0.22) ( 2 C 2p+1: -0.52) ( 2 C 2p-1: 0.57)
( 3 O 3s : -0.48) ( 3 O 2p-1: -0.19) ( 4 H 2s : 0.98)
( 5 H 2s : -0.83) ( 6 H 2s : -0.31) ( 7 H 2s : 0.83)
( 8 H 2s : -1.35) ( 9 H 2s : 0.51)
Molecular Orbital No. 18:
--------------------------
Occupation: 0.000 Energy: 0.13483 a.u.
( 1 C 3s : -0.53) ( 1 C 1p+1: -0.23) ( 1 C 1p0 : -0.22)
( 1 C 2p+1: -0.85) ( 1 C 2p-1: -0.51) ( 1 C 2p0 : -0.67)
( 2 C 3s : 0.95) ( 2 C 2p+1: 0.30) ( 2 C 2p-1: 0.23)
( 4 H 2s : -0.61) ( 5 H 2s : -0.66) ( 6 H 2s : 1.94)
( 7 H 2s : -0.59) ( 8 H 2s : -0.64) ( 9 H 2s : 0.21)
Ground State Dipole Moment
----------------------------
X : -0.242040 a.u. -0.615205 Debye
Y : 0.403846 a.u. 1.026473 Debye
Z : 0.395932 a.u. 1.006358 Debye
Total : 0.615172 a.u. 1.563611 Debye
SCF Gradient Driver
=====================
Gradient Type : Analytical
Molecular Geometry (Angstroms)
================================
Atom Coordinate X Coordinate Y Coordinate Z
C 0.552463000000 0.063946000000 0.841471000000
C 1.085653000000 0.776448000000 -0.395425000000
O 0.354459000000 0.397756000000 -1.529505000000
H 0.613582000000 -1.036253000000 0.702300000000
H -0.505156000000 0.351308000000 1.021250000000
H 1.157624000000 0.349252000000 1.727669000000
H 2.149329000000 0.495326000000 -0.545921000000
H 1.037532000000 1.879437000000 -0.249306000000
H -0.508089000000 0.887808000000 -1.487835000000
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.004456948769 -0.001323207600 -0.005937503739
C 0.000977253166 -0.000244809361 -0.008590980002
O 0.021578125998 -0.012609790265 0.011283222002
H 0.001751932931 -0.006202137369 -0.001446049631
H -0.004522607853 0.002809968113 -0.001042576740
H 0.004798934659 0.002845088464 0.001181333112
H 0.004781290859 -0.002260418264 0.003025189629
H -0.004369175727 0.002676433528 -0.000863608739
H -0.020576210272 0.014325777029 0.002382986978
*** Time spent in gradient calculation: 1.00 sec ***
* Info * Energy : -154.9215318607 a.u.
* Info * Gradient : 1.369353e-02 a.u. (RMS)
* Info * 2.742140e-02 a.u. (Max)
* Info * Time : 2.31 sec
* Info * Computing energy and gradient...
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Kohn-Sham
Initial Guess Model : Restart from Checkpoint
Convergence Accelerator : Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Nuclear repulsion energy: 81.2790878917 a.u.
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Molecular grid with 115509 points generated in 0.04 sec.
* Info * Restarting from checkpoint file: vlx_20251015_6d4463a8_scf.h5
* Info * Overlap matrix computed in 0.00 sec.
* Info * Kinetic energy matrix computed in 0.01 sec.
* Info * Nuclear potential matrix computed in 0.00 sec.
* Info * Orthogonalization matrix computed in 0.00 sec.
Iter. | Kohn-Sham Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -154.935346810365 0.0000000000 0.21272222 0.02433727 0.00000000
2 -154.923291266684 0.0120555437 0.03518479 0.00205522 0.06572674
3 -154.923350789546 -0.0000595229 0.02317451 0.00118019 0.01884868
4 -154.923405608162 -0.0000548186 0.00726700 0.00040855 0.00912246
5 -154.923411983896 -0.0000063757 0.00070102 0.00002977 0.00224908
6 -154.923412024261 -0.0000000404 0.00038833 0.00002516 0.00029984
7 -154.923412037885 -0.0000000136 0.00009680 0.00000650 0.00013063
8 -154.923412038850 -0.0000000010 0.00000957 0.00000069 0.00003418
9 -154.923412038861 -0.0000000000 0.00000160 0.00000009 0.00000452
10 -154.923412038861 -0.0000000000 0.00000065 0.00000004 0.00000076
* Info * Checkpoint written to file: vlx_20251015_6d4463a8_scf.h5
*** SCF converged in 10 iterations. Time: 1.10 sec.
Spin-Restricted Kohn-Sham:
--------------------------
Total Energy : -154.9234120389 a.u.
Electronic Energy : -236.2024999306 a.u.
Nuclear Repulsion Energy : 81.2790878917 a.u.
------------------------------------
Gradient Norm : 0.0000006543 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
Spin Restricted Orbitals
------------------------
Molecular Orbital No. 9:
--------------------------
Occupation: 2.000 Energy: -0.42292 a.u.
( 1 C 1p0 : -0.35) ( 2 C 1p-1: -0.22) ( 2 C 1p0 : 0.21)
( 3 O 1p+1: -0.23) ( 6 H 1s : -0.22)
Molecular Orbital No. 10:
--------------------------
Occupation: 2.000 Energy: -0.38135 a.u.
( 1 C 1p-1: -0.38) ( 1 C 2p-1: -0.17) ( 3 O 1p-1: 0.22)
( 4 H 1s : 0.33) ( 6 H 1s : -0.17)
Molecular Orbital No. 11:
--------------------------
Occupation: 2.000 Energy: -0.36875 a.u.
( 1 C 1p+1: 0.33) ( 2 C 1p+1: -0.21) ( 3 O 1p-1: 0.15)
( 3 O 1p0 : 0.26) ( 3 O 2p0 : 0.16) ( 5 H 1s : -0.29)
( 6 H 1s : 0.22) ( 7 H 1s : -0.18)
Molecular Orbital No. 12:
--------------------------
Occupation: 2.000 Energy: -0.31946 a.u.
( 2 C 1p+1: -0.15) ( 2 C 1p-1: 0.20) ( 2 C 1p0 : 0.18)
( 3 O 3s : 0.21) ( 3 O 1p+1: 0.16) ( 3 O 1p-1: -0.23)
( 3 O 1p0 : -0.28) ( 3 O 2p-1: -0.15) ( 3 O 2p0 : -0.20)
( 7 H 1s : -0.26) ( 7 H 2s : -0.16) ( 8 H 1s : 0.24)
( 8 H 2s : 0.16) ( 9 H 1s : -0.19)
Molecular Orbital No. 13:
--------------------------
Occupation: 2.000 Energy: -0.26849 a.u.
( 1 C 3s : -0.20) ( 1 C 1p0 : 0.20) ( 3 O 1p+1: -0.22)
( 3 O 1p-1: -0.45) ( 3 O 1p0 : 0.28) ( 3 O 2p+1: -0.16)
( 3 O 2p-1: -0.34) ( 3 O 2p0 : 0.22) ( 8 H 1s : 0.17)
Molecular Orbital No. 14:
--------------------------
Occupation: 0.000 Energy: 0.04774 a.u.
( 1 C 3s : 1.07) ( 1 C 2p+1: -0.25) ( 1 C 2p0 : 0.26)
( 2 C 3s : 1.01) ( 2 C 2p+1: 0.21) ( 2 C 2p-1: 0.34)
( 3 O 3s : 0.55) ( 3 O 1p+1: -0.18) ( 3 O 2p+1: -0.27)
( 4 H 2s : -0.41) ( 5 H 2s : -0.71) ( 6 H 2s : -0.54)
( 7 H 2s : -0.41) ( 8 H 2s : -0.81) ( 9 H 2s : -0.90)
Molecular Orbital No. 15:
--------------------------
Occupation: 0.000 Energy: 0.08747 a.u.
( 1 C 3s : -1.87) ( 1 C 2p-1: 0.17) ( 1 C 2p0 : -0.25)
( 2 C 3s : -0.48) ( 2 C 2p+1: -0.36) ( 2 C 2p0 : 0.35)
( 3 O 3s : 0.62) ( 3 O 2p-1: 0.17) ( 4 H 2s : 0.94)
( 5 H 2s : 0.50) ( 6 H 2s : 1.02) ( 7 H 2s : 0.89)
( 9 H 2s : -0.85)
Molecular Orbital No. 16:
--------------------------
Occupation: 0.000 Energy: 0.10763 a.u.
( 1 C 3s : 2.17) ( 1 C 1p+1: -0.15) ( 1 C 2p+1: -0.31)
( 1 C 2p0 : -0.27) ( 2 C 3s : -2.20) ( 2 C 1p+1: -0.16)
( 2 C 2p+1: -0.36) ( 2 C 2p0 : -0.39) ( 4 H 2s : -0.77)
( 5 H 2s : -1.33) ( 7 H 2s : 1.30) ( 8 H 2s : 1.06)
Molecular Orbital No. 17:
--------------------------
Occupation: 0.000 Energy: 0.12027 a.u.
( 1 C 1p-1: 0.17) ( 1 C 2p+1: -0.31) ( 1 C 2p-1: 0.41)
( 1 C 2p0 : 0.18) ( 2 C 3s : 0.68) ( 2 C 1p+1: -0.19)
( 2 C 1p-1: 0.23) ( 2 C 2p+1: -0.50) ( 2 C 2p-1: 0.62)
( 3 O 3s : -0.46) ( 3 O 2p-1: -0.17) ( 4 H 2s : 0.91)
( 5 H 2s : -0.81) ( 7 H 2s : 0.79) ( 8 H 2s : -1.49)
( 9 H 2s : 0.46)
Molecular Orbital No. 18:
--------------------------
Occupation: 0.000 Energy: 0.13413 a.u.
( 1 C 3s : -0.56) ( 1 C 1p+1: -0.22) ( 1 C 1p-1: -0.16)
( 1 C 1p0 : -0.21) ( 1 C 2p+1: -0.82) ( 1 C 2p-1: -0.61)
( 1 C 2p0 : -0.64) ( 2 C 3s : 0.96) ( 2 C 2p+1: 0.31)
( 4 H 2s : -0.80) ( 5 H 2s : -0.53) ( 6 H 2s : 1.96)
( 7 H 2s : -0.67) ( 8 H 2s : -0.45)
Ground State Dipole Moment
----------------------------
X : -0.259777 a.u. -0.660288 Debye
Y : 0.374249 a.u. 0.951246 Debye
Z : 0.421314 a.u. 1.070874 Debye
Total : 0.620526 a.u. 1.577219 Debye
SCF Gradient Driver
=====================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
================================
Atom Coordinate X Coordinate Y Coordinate Z
C 0.557953886851 0.067349665838 0.862418475147
C 1.066714632861 0.780801507500 -0.393124832058
O 0.365625389885 0.421060585709 -1.569064705281
H 0.600110940743 -1.022746762002 0.719732087140
H -0.488077960783 0.345181399804 1.073543132976
H 1.160626531052 0.325803290083 1.749838654445
H 2.119249227133 0.506151895460 -0.574077253717
H 1.052306097389 1.877762934526 -0.229503462608
H -0.493548847156 0.862053318868 -1.554303306774
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000285607044 -0.001102397816 0.003471669488
C -0.000546528862 0.000809443665 -0.002012380075
O -0.000254774610 -0.001221297552 -0.001332497928
H 0.000548409956 0.000828791018 -0.000278410826
H 0.000597893876 0.000228105221 -0.000480675901
H -0.000081028735 -0.000496614166 0.001013463374
H -0.000170902118 0.000008866723 -0.000330178266
H -0.000199653887 -0.000137264709 0.000597162522
H 0.000383413977 0.001090302266 -0.000654870963
*** Time spent in gradient calculation: 1.09 sec ***
* Info * Energy : -154.9234120389 a.u.
* Info * Gradient : 1.731704e-03 a.u. (RMS)
* Info * 3.653675e-03 a.u. (Max)
* Info * Time : 2.26 sec
* Info * Computing energy and gradient...
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Kohn-Sham
Initial Guess Model : Restart from Checkpoint
Convergence Accelerator : Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Nuclear repulsion energy: 81.4346862042 a.u.
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Molecular grid with 115465 points generated in 0.04 sec.
* Info * Restarting from checkpoint file: vlx_20251015_6d4463a8_scf.h5
* Info * Overlap matrix computed in 0.00 sec.
* Info * Kinetic energy matrix computed in 0.00 sec.
* Info * Nuclear potential matrix computed in 0.01 sec.
* Info * Orthogonalization matrix computed in 0.00 sec.
Iter. | Kohn-Sham Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -154.927044928799 0.0000000000 0.04011315 0.00337006 0.00000000
2 -154.923527215160 0.0035177136 0.00871562 0.00080742 0.02353782
3 -154.923528635649 -0.0000014205 0.00756767 0.00044881 0.00496536
4 -154.923534522566 -0.0000058869 0.00241996 0.00016011 0.00267457
5 -154.923535093499 -0.0000005709 0.00057703 0.00003913 0.00077256
6 -154.923535121340 -0.0000000278 0.00019898 0.00001102 0.00024125
7 -154.923535125722 -0.0000000044 0.00000975 0.00000059 0.00007206
8 -154.923535125737 -0.0000000000 0.00000236 0.00000021 0.00000644
9 -154.923535125738 -0.0000000000 0.00000040 0.00000003 0.00000132
* Info * Checkpoint written to file: vlx_20251015_6d4463a8_scf.h5
*** SCF converged in 9 iterations. Time: 1.04 sec.
Spin-Restricted Kohn-Sham:
--------------------------
Total Energy : -154.9235351257 a.u.
Electronic Energy : -236.3582213300 a.u.
Nuclear Repulsion Energy : 81.4346862042 a.u.
------------------------------------
Gradient Norm : 0.0000003981 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
Spin Restricted Orbitals
------------------------
Molecular Orbital No. 9:
--------------------------
Occupation: 2.000 Energy: -0.42396 a.u.
( 1 C 1p0 : -0.35) ( 2 C 1p-1: -0.23) ( 2 C 1p0 : 0.21)
( 3 O 1p+1: -0.23) ( 6 H 1s : -0.22)
Molecular Orbital No. 10:
--------------------------
Occupation: 2.000 Energy: -0.38174 a.u.
( 1 C 1p-1: -0.37) ( 1 C 2p-1: -0.17) ( 3 O 1p-1: 0.20)
( 4 H 1s : 0.33) ( 6 H 1s : -0.18)
Molecular Orbital No. 11:
--------------------------
Occupation: 2.000 Energy: -0.36859 a.u.
( 1 C 1p+1: 0.32) ( 2 C 1p+1: -0.21) ( 3 O 1p-1: 0.17)
( 3 O 1p0 : 0.25) ( 3 O 2p0 : 0.16) ( 5 H 1s : -0.29)
( 6 H 1s : 0.21) ( 7 H 1s : -0.18)
Molecular Orbital No. 12:
--------------------------
Occupation: 2.000 Energy: -0.31931 a.u.
( 2 C 1p+1: -0.15) ( 2 C 1p-1: 0.19) ( 2 C 1p0 : 0.18)
( 3 O 3s : 0.21) ( 3 O 1p+1: 0.17) ( 3 O 1p-1: -0.23)
( 3 O 1p0 : -0.28) ( 3 O 2p0 : -0.21) ( 7 H 1s : -0.26)
( 7 H 2s : -0.16) ( 8 H 1s : 0.23) ( 8 H 2s : 0.15)
( 9 H 1s : -0.19)
Molecular Orbital No. 13:
--------------------------
Occupation: 2.000 Energy: -0.26823 a.u.
( 1 C 3s : -0.19) ( 1 C 1p0 : 0.19) ( 3 O 1p+1: -0.21)
( 3 O 1p-1: -0.46) ( 3 O 1p0 : 0.28) ( 3 O 2p+1: -0.15)
( 3 O 2p-1: -0.35) ( 3 O 2p0 : 0.22) ( 8 H 1s : 0.18)
Molecular Orbital No. 14:
--------------------------
Occupation: 0.000 Energy: 0.04813 a.u.
( 1 C 3s : 1.10) ( 1 C 2p+1: -0.26) ( 1 C 2p0 : 0.26)
( 2 C 3s : 1.00) ( 2 C 2p+1: 0.21) ( 2 C 2p-1: 0.33)
( 3 O 3s : 0.54) ( 3 O 1p+1: -0.18) ( 3 O 2p+1: -0.27)
( 4 H 2s : -0.42) ( 5 H 2s : -0.73) ( 6 H 2s : -0.55)
( 7 H 2s : -0.42) ( 8 H 2s : -0.81) ( 9 H 2s : -0.90)
Molecular Orbital No. 15:
--------------------------
Occupation: 0.000 Energy: 0.08767 a.u.
( 1 C 3s : -1.84) ( 1 C 2p-1: 0.17) ( 1 C 2p0 : -0.25)
( 2 C 3s : -0.49) ( 2 C 2p+1: -0.37) ( 2 C 2p0 : 0.35)
( 3 O 3s : 0.62) ( 3 O 2p-1: 0.16) ( 4 H 2s : 0.93)
( 5 H 2s : 0.47) ( 6 H 2s : 1.01) ( 7 H 2s : 0.90)
( 9 H 2s : -0.85)
Molecular Orbital No. 16:
--------------------------
Occupation: 0.000 Energy: 0.10701 a.u.
( 1 C 3s : 2.19) ( 1 C 1p+1: -0.15) ( 1 C 2p+1: -0.31)
( 1 C 2p0 : -0.28) ( 2 C 3s : -2.19) ( 2 C 1p+1: -0.16)
( 2 C 2p+1: -0.35) ( 2 C 2p0 : -0.39) ( 4 H 2s : -0.80)
( 5 H 2s : -1.33) ( 7 H 2s : 1.28) ( 8 H 2s : 1.06)
Molecular Orbital No. 17:
--------------------------
Occupation: 0.000 Energy: 0.11994 a.u.
( 1 C 1p-1: 0.17) ( 1 C 2p+1: -0.29) ( 1 C 2p-1: 0.43)
( 1 C 2p0 : 0.19) ( 2 C 3s : 0.64) ( 2 C 1p+1: -0.18)
( 2 C 1p-1: 0.23) ( 2 C 2p+1: -0.49) ( 2 C 2p-1: 0.61)
( 3 O 3s : -0.46) ( 3 O 2p-1: -0.17) ( 4 H 2s : 0.93)
( 5 H 2s : -0.82) ( 7 H 2s : 0.79) ( 8 H 2s : -1.47)
( 9 H 2s : 0.46)
Molecular Orbital No. 18:
--------------------------
Occupation: 0.000 Energy: 0.13467 a.u.
( 1 C 3s : -0.57) ( 1 C 1p+1: -0.22) ( 1 C 1p-1: -0.16)
( 1 C 1p0 : -0.21) ( 1 C 2p+1: -0.83) ( 1 C 2p-1: -0.60)
( 1 C 2p0 : -0.65) ( 2 C 3s : 0.98) ( 2 C 2p+1: 0.32)
( 4 H 2s : -0.78) ( 5 H 2s : -0.53) ( 6 H 2s : 1.97)
( 7 H 2s : -0.68) ( 8 H 2s : -0.50)
Ground State Dipole Moment
----------------------------
X : -0.262462 a.u. -0.667112 Debye
Y : 0.359607 a.u. 0.914029 Debye
Z : 0.430941 a.u. 1.095344 Debye
Total : 0.619608 a.u. 1.574886 Debye
SCF Gradient Driver
=====================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
================================
Atom Coordinate X Coordinate Y Coordinate Z
C 0.555650521629 0.068808778350 0.854360457949
C 1.072578451028 0.783720644833 -0.391223159840
O 0.368574555064 0.424812998814 -1.564216446472
H 0.591728682167 -1.022463529351 0.713227697052
H -0.490218055163 0.348455650237 1.067050365886
H 1.158749769555 0.326763327735 1.740066353530
H 2.125084992989 0.508654098490 -0.570887135690
H 1.059815088512 1.880639697855 -0.227887263674
H -0.500087430039 0.843260112526 -1.534406852423
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000305806459 -0.000487759143 0.000940900371
C 0.000666696463 0.000803451002 -0.000590120326
O -0.001650009615 -0.000720622677 0.000644658954
H 0.000101068842 0.000238647105 0.000014782062
H 0.000226166746 -0.000119007631 -0.000136103894
H -0.000119583001 -0.000095530608 -0.000329143401
H -0.000096456875 0.000288279424 -0.000117876163
H -0.000160286445 -0.000295200614 0.000268308157
H 0.001328931004 0.000391812372 -0.000700422150
*** Time spent in gradient calculation: 0.99 sec ***
* Info * Energy : -154.9235351257 a.u.
* Info * Gradient : 1.016348e-03 a.u. (RMS)
* Info * 1.912437e-03 a.u. (Max)
* Info * Time : 2.11 sec
* Info * Computing energy and gradient...
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Kohn-Sham
Initial Guess Model : Restart from Checkpoint
Convergence Accelerator : Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Nuclear repulsion energy: 81.5335514112 a.u.
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Molecular grid with 115461 points generated in 0.04 sec.
* Info * Restarting from checkpoint file: vlx_20251015_6d4463a8_scf.h5
* Info * Overlap matrix computed in 0.00 sec.
* Info * Kinetic energy matrix computed in 0.00 sec.
* Info * Nuclear potential matrix computed in 0.00 sec.
* Info * Orthogonalization matrix computed in 0.00 sec.
Iter. | Kohn-Sham Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -154.924595445220 0.0000000000 0.06293716 0.00432073 0.00000000
2 -154.923611168795 0.0009842764 0.01612965 0.00169573 0.03951510
3 -154.923621746014 -0.0000105772 0.01126253 0.00074134 0.00776553
4 -154.923630900820 -0.0000091548 0.00585447 0.00039546 0.00449925
5 -154.923634173487 -0.0000032727 0.00053626 0.00003005 0.00167439
6 -154.923634192927 -0.0000000194 0.00029620 0.00001793 0.00026226
7 -154.923634203694 -0.0000000108 0.00002606 0.00000173 0.00010625
8 -154.923634203793 -0.0000000001 0.00000595 0.00000040 0.00001471
9 -154.923634203799 -0.0000000000 0.00000067 0.00000006 0.00000323
* Info * Checkpoint written to file: vlx_20251015_6d4463a8_scf.h5
*** SCF converged in 9 iterations. Time: 0.93 sec.
Spin-Restricted Kohn-Sham:
--------------------------
Total Energy : -154.9236342038 a.u.
Electronic Energy : -236.4571856150 a.u.
Nuclear Repulsion Energy : 81.5335514112 a.u.
------------------------------------
Gradient Norm : 0.0000006656 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
Spin Restricted Orbitals
------------------------
Molecular Orbital No. 9:
--------------------------
Occupation: 2.000 Energy: -0.42611 a.u.
( 1 C 1p0 : -0.35) ( 2 C 1p-1: -0.23) ( 2 C 1p0 : 0.20)
( 3 O 1p+1: -0.22) ( 6 H 1s : -0.21)
Molecular Orbital No. 10:
--------------------------
Occupation: 2.000 Energy: -0.38172 a.u.
( 1 C 1p-1: -0.37) ( 1 C 2p-1: -0.17) ( 2 C 1p-1: 0.15)
( 3 O 1p-1: 0.19) ( 4 H 1s : 0.32) ( 6 H 1s : -0.19)
Molecular Orbital No. 11:
--------------------------
Occupation: 2.000 Energy: -0.36853 a.u.
( 1 C 1p+1: 0.32) ( 2 C 1p+1: -0.20) ( 3 O 1p-1: 0.18)
( 3 O 1p0 : 0.25) ( 3 O 2p0 : 0.16) ( 5 H 1s : -0.29)
( 6 H 1s : 0.21) ( 7 H 1s : -0.18)
Molecular Orbital No. 12:
--------------------------
Occupation: 2.000 Energy: -0.31946 a.u.
( 2 C 1p+1: -0.15) ( 2 C 1p-1: 0.18) ( 2 C 1p0 : 0.19)
( 3 O 3s : 0.21) ( 3 O 1p+1: 0.17) ( 3 O 1p-1: -0.20)
( 3 O 1p0 : -0.30) ( 3 O 2p0 : -0.22) ( 7 H 1s : -0.26)
( 7 H 2s : -0.16) ( 8 H 1s : 0.22) ( 9 H 1s : -0.19)
Molecular Orbital No. 13:
--------------------------
Occupation: 2.000 Energy: -0.26809 a.u.
( 1 C 3s : -0.18) ( 1 C 1p0 : 0.19) ( 2 C 1p-1: 0.16)
( 3 O 1p+1: -0.18) ( 3 O 1p-1: -0.48) ( 3 O 1p0 : 0.26)
( 3 O 2p-1: -0.36) ( 3 O 2p0 : 0.21) ( 8 H 1s : 0.19)
Molecular Orbital No. 14:
--------------------------
Occupation: 0.000 Energy: 0.04835 a.u.
( 1 C 3s : 1.14) ( 1 C 2p+1: -0.27) ( 1 C 2p0 : 0.27)
( 2 C 3s : 0.99) ( 2 C 2p+1: 0.21) ( 2 C 2p-1: 0.32)
( 3 O 3s : 0.53) ( 3 O 1p+1: -0.18) ( 3 O 2p+1: -0.28)
( 4 H 2s : -0.43) ( 5 H 2s : -0.76) ( 6 H 2s : -0.56)
( 7 H 2s : -0.41) ( 8 H 2s : -0.78) ( 9 H 2s : -0.89)
Molecular Orbital No. 15:
--------------------------
Occupation: 0.000 Energy: 0.08783 a.u.
( 1 C 3s : -1.79) ( 1 C 2p-1: 0.17) ( 1 C 2p0 : -0.25)
( 2 C 3s : -0.53) ( 2 C 2p+1: -0.38) ( 2 C 2p0 : 0.35)
( 3 O 3s : 0.63) ( 3 O 2p-1: 0.15) ( 4 H 2s : 0.92)
( 5 H 2s : 0.41) ( 6 H 2s : 1.01) ( 7 H 2s : 0.93)
( 8 H 2s : 0.16) ( 9 H 2s : -0.87)
Molecular Orbital No. 16:
--------------------------
Occupation: 0.000 Energy: 0.10637 a.u.
( 1 C 3s : 2.22) ( 1 C 2p+1: -0.30) ( 1 C 2p0 : -0.28)
( 2 C 3s : -2.18) ( 2 C 1p+1: -0.15) ( 2 C 2p+1: -0.34)
( 2 C 2p0 : -0.41) ( 4 H 2s : -0.84) ( 5 H 2s : -1.32)
( 7 H 2s : 1.25) ( 8 H 2s : 1.07)
Molecular Orbital No. 17:
--------------------------
Occupation: 0.000 Energy: 0.11932 a.u.
( 1 C 1p-1: 0.18) ( 1 C 2p+1: -0.30) ( 1 C 2p-1: 0.44)
( 1 C 2p0 : 0.18) ( 2 C 3s : 0.61) ( 2 C 1p+1: -0.18)
( 2 C 1p-1: 0.23) ( 2 C 2p+1: -0.47) ( 2 C 2p-1: 0.62)
( 3 O 3s : -0.45) ( 3 O 2p-1: -0.16) ( 4 H 2s : 0.93)
( 5 H 2s : -0.85) ( 7 H 2s : 0.79) ( 8 H 2s : -1.45)
( 9 H 2s : 0.47)
Molecular Orbital No. 18:
--------------------------
Occupation: 0.000 Energy: 0.13445 a.u.
( 1 C 3s : -0.59) ( 1 C 1p+1: -0.21) ( 1 C 1p-1: -0.16)
( 1 C 1p0 : -0.22) ( 1 C 2p+1: -0.83) ( 1 C 2p-1: -0.59)
( 1 C 2p0 : -0.66) ( 2 C 3s : 0.99) ( 2 C 2p+1: 0.33)
( 2 C 2p-1: 0.15) ( 4 H 2s : -0.78) ( 5 H 2s : -0.52)
( 6 H 2s : 1.97) ( 7 H 2s : -0.69) ( 8 H 2s : -0.51)
Ground State Dipole Moment
----------------------------
X : -0.270827 a.u. -0.688374 Debye
Y : 0.327902 a.u. 0.833444 Debye
Z : 0.451892 a.u. 1.148594 Debye
Total : 0.620543 a.u. 1.577262 Debye
SCF Gradient Driver
=====================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
================================
Atom Coordinate X Coordinate Y Coordinate Z
C 0.553720728043 0.071494404324 0.846247630811
C 1.078883568567 0.787630895342 -0.390169254717
O 0.376270101155 0.436002434842 -1.565253115545
H 0.580663579797 -1.020555193652 0.703487216744
H -0.491403283393 0.356508365640 1.059677080098
H 1.155838130040 0.323942671971 1.734317933910
H 2.131175593052 0.510306635528 -0.568245473041
H 1.069719689789 1.884787037160 -0.224863032567
H -0.511006623273 0.811476699992 -1.508006482785
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000157632229 0.000307292378 -0.001351884905
C 0.000648390782 -0.000042368240 0.000861606722
O -0.001305885466 -0.000573285618 0.001353510024
H -0.000224108039 -0.000237321489 0.000137762706
H -0.000147888326 -0.000245383282 0.000095838998
H 0.000038872778 0.000212308564 -0.000542388979
H 0.000076792017 0.000398145777 -0.000003773466
H -0.000169041018 -0.000173315319 -0.000059624403
H 0.000916950035 0.000359029546 -0.000497024541
*** Time spent in gradient calculation: 0.92 sec ***
* Info * Energy : -154.9236342038 a.u.
* Info * Gradient : 9.987949e-04 a.u. (RMS)
* Info * 1.966210e-03 a.u. (Max)
* Info * Time : 1.92 sec
* Info * Computing energy and gradient...
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Kohn-Sham
Initial Guess Model : Restart from Checkpoint
Convergence Accelerator : Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Nuclear repulsion energy: 81.5786436603 a.u.
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Molecular grid with 115419 points generated in 0.04 sec.
* Info * Restarting from checkpoint file: vlx_20251015_6d4463a8_scf.h5
* Info * Overlap matrix computed in 0.00 sec.
* Info * Kinetic energy matrix computed in 0.00 sec.
* Info * Nuclear potential matrix computed in 0.00 sec.
* Info * Orthogonalization matrix computed in 0.00 sec.
Iter. | Kohn-Sham Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -154.921886071389 0.0000000000 0.10265993 0.00747599 0.00000000
2 -154.923664019294 -0.0017779479 0.02753026 0.00292647 0.06521211
3 -154.923692849844 -0.0000288306 0.02004831 0.00143327 0.01390531
4 -154.923720612159 -0.0000277623 0.01020637 0.00076523 0.00825968
5 -154.923730142177 -0.0000095300 0.00072222 0.00003972 0.00289460
6 -154.923730185778 -0.0000000436 0.00034295 0.00001864 0.00032443
7 -154.923730199854 -0.0000000141 0.00005496 0.00000309 0.00011450
8 -154.923730200250 -0.0000000004 0.00001112 0.00000074 0.00002578
9 -154.923730200268 -0.0000000000 0.00000111 0.00000008 0.00000573
10 -154.923730200268 -0.0000000000 0.00000021 0.00000002 0.00000063
* Info * Checkpoint written to file: vlx_20251015_6d4463a8_scf.h5
*** SCF converged in 10 iterations. Time: 1.08 sec.
Spin-Restricted Kohn-Sham:
--------------------------
Total Energy : -154.9237302003 a.u.
Electronic Energy : -236.5023738606 a.u.
Nuclear Repulsion Energy : 81.5786436603 a.u.
------------------------------------
Gradient Norm : 0.0000002123 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
Spin Restricted Orbitals
------------------------
Molecular Orbital No. 9:
--------------------------
Occupation: 2.000 Energy: -0.42944 a.u.
( 1 C 1p0 : -0.34) ( 1 C 2p0 : -0.15) ( 2 C 1p-1: -0.24)
( 2 C 1p0 : 0.18) ( 3 O 1p+1: -0.20) ( 6 H 1s : -0.22)
Molecular Orbital No. 10:
--------------------------
Occupation: 2.000 Energy: -0.38122 a.u.
( 1 C 1p-1: -0.37) ( 1 C 2p-1: -0.16) ( 2 C 1p-1: 0.16)
( 3 O 1p-1: 0.18) ( 4 H 1s : 0.32) ( 6 H 1s : -0.19)
Molecular Orbital No. 11:
--------------------------
Occupation: 2.000 Energy: -0.36881 a.u.
( 1 C 1p+1: 0.31) ( 2 C 1p+1: -0.20) ( 3 O 1p-1: 0.18)
( 3 O 1p0 : 0.25) ( 3 O 2p0 : 0.15) ( 5 H 1s : -0.29)
( 6 H 1s : 0.20) ( 7 H 1s : -0.18)
Molecular Orbital No. 12:
--------------------------
Occupation: 2.000 Energy: -0.32017 a.u.
( 1 C 1p0 : -0.17) ( 2 C 1p+1: -0.16) ( 2 C 1p-1: 0.16)
( 2 C 1p0 : 0.21) ( 3 O 3s : 0.21) ( 3 O 1p+1: 0.19)
( 3 O 1p-1: -0.16) ( 3 O 1p0 : -0.32) ( 3 O 2p0 : -0.23)
( 7 H 1s : -0.26) ( 7 H 2s : -0.16) ( 8 H 1s : 0.19)
( 9 H 1s : -0.19)
Molecular Orbital No. 13:
--------------------------
Occupation: 2.000 Energy: -0.26789 a.u.
( 1 C 3s : -0.16) ( 1 C 1p0 : 0.18) ( 2 C 1p-1: 0.17)
( 3 O 1p-1: -0.51) ( 3 O 1p0 : 0.23) ( 3 O 2p-1: -0.38)
( 3 O 2p0 : 0.19) ( 8 H 1s : 0.21)
Molecular Orbital No. 14:
--------------------------
Occupation: 0.000 Energy: 0.04872 a.u.
( 1 C 3s : 1.21) ( 1 C 2p+1: -0.29) ( 1 C 2p0 : 0.28)
( 2 C 3s : 0.95) ( 2 C 2p+1: 0.21) ( 2 C 2p-1: 0.29)
( 3 O 3s : 0.52) ( 3 O 1p+1: -0.18) ( 3 O 2p+1: -0.28)
( 4 H 2s : -0.46) ( 5 H 2s : -0.80) ( 6 H 2s : -0.59)
( 7 H 2s : -0.42) ( 8 H 2s : -0.74) ( 9 H 2s : -0.88)
Molecular Orbital No. 15:
--------------------------
Occupation: 0.000 Energy: 0.08807 a.u.
( 1 C 3s : -1.69) ( 1 C 2p+1: -0.15) ( 1 C 2p-1: 0.17)
( 1 C 2p0 : -0.24) ( 2 C 3s : -0.59) ( 2 C 2p+1: -0.39)
( 2 C 2p0 : 0.34) ( 3 O 3s : 0.64) ( 3 O 2p+1: -0.16)
( 4 H 2s : 0.88) ( 5 H 2s : 0.34) ( 6 H 2s : 0.98)
( 7 H 2s : 0.95) ( 8 H 2s : 0.22) ( 9 H 2s : -0.89)
Molecular Orbital No. 16:
--------------------------
Occupation: 0.000 Energy: 0.10581 a.u.
( 1 C 3s : 2.25) ( 1 C 2p+1: -0.29) ( 1 C 2p0 : -0.28)
( 2 C 3s : -2.17) ( 2 C 1p+1: -0.15) ( 2 C 2p+1: -0.33)
( 2 C 2p0 : -0.43) ( 4 H 2s : -0.90) ( 5 H 2s : -1.30)
( 7 H 2s : 1.20) ( 8 H 2s : 1.09)
Molecular Orbital No. 17:
--------------------------
Occupation: 0.000 Energy: 0.11834 a.u.
( 1 C 1p-1: 0.18) ( 1 C 2p+1: -0.33) ( 1 C 2p-1: 0.45)
( 1 C 2p0 : 0.15) ( 2 C 3s : 0.56) ( 2 C 1p+1: -0.18)
( 2 C 1p-1: 0.23) ( 2 C 2p+1: -0.44) ( 2 C 2p-1: 0.63)
( 3 O 3s : -0.43) ( 4 H 2s : 0.92) ( 5 H 2s : -0.90)
( 7 H 2s : 0.78) ( 8 H 2s : -1.43) ( 9 H 2s : 0.45)
Molecular Orbital No. 18:
--------------------------
Occupation: 0.000 Energy: 0.13376 a.u.
( 1 C 3s : -0.61) ( 1 C 1p+1: -0.21) ( 1 C 1p-1: -0.16)
( 1 C 1p0 : -0.22) ( 1 C 2p+1: -0.82) ( 1 C 2p-1: -0.59)
( 1 C 2p0 : -0.67) ( 2 C 3s : 1.00) ( 2 C 2p+1: 0.35)
( 4 H 2s : -0.79) ( 5 H 2s : -0.48) ( 6 H 2s : 1.98)
( 7 H 2s : -0.72) ( 8 H 2s : -0.50)
Ground State Dipole Moment
----------------------------
X : -0.283747 a.u. -0.721214 Debye
Y : 0.271407 a.u. 0.689848 Debye
Z : 0.485408 a.u. 1.233785 Debye
Total : 0.624336 a.u. 1.586904 Debye
SCF Gradient Driver
=====================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
================================
Atom Coordinate X Coordinate Y Coordinate Z
C 0.550826252957 0.075780413104 0.837628926029
C 1.086493292122 0.795228320133 -0.390072327008
O 0.388254111010 0.457792365460 -1.571451250158
H 0.564161281352 -1.016005177659 0.688355115463
H -0.491786803409 0.371125385053 1.052788401480
H 1.151772752699 0.315168667461 1.730294414631
H 2.137317314215 0.511569420012 -0.566929467838
H 1.085485414558 1.892153793009 -0.217846063558
H -0.525098053770 0.756511593434 -1.473230393708
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000640223538 0.000774288178 -0.002340402573
C 0.000127900801 -0.000673249636 0.001700607146
O -0.000432712143 -0.000258304454 0.000908039567
H -0.000422124007 -0.000415216827 0.000199188859
H -0.000347738249 -0.000255650786 0.000287573857
H 0.000040248564 0.000289031727 -0.000382109399
H 0.000109456874 0.000337817562 -0.000065091686
H -0.000012159226 0.000061741417 -0.000241543131
H 0.000290182693 0.000149530609 -0.000079575760
*** Time spent in gradient calculation: 0.98 sec ***
* Info * Energy : -154.9237302003 a.u.
* Info * Gradient : 1.160605e-03 a.u. (RMS)
* Info * 2.546938e-03 a.u. (Max)
* Info * Time : 2.13 sec
* Info * Computing energy and gradient...
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Kohn-Sham
Initial Guess Model : Restart from Checkpoint
Convergence Accelerator : Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Nuclear repulsion energy: 81.5389082615 a.u.
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Molecular grid with 115411 points generated in 0.07 sec.
* Info * Restarting from checkpoint file: vlx_20251015_6d4463a8_scf.h5
* Info * Overlap matrix computed in 0.00 sec.
* Info * Kinetic energy matrix computed in 0.00 sec.
* Info * Nuclear potential matrix computed in 0.00 sec.
* Info * Orthogonalization matrix computed in 0.00 sec.
Iter. | Kohn-Sham Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -154.922207458514 0.0000000000 0.03715657 0.00270324 0.00000000
2 -154.923746780461 -0.0015393219 0.00901696 0.00102877 0.02249624
3 -154.923752151618 -0.0000053712 0.00399584 0.00036466 0.00412877
4 -154.923753372588 -0.0000012210 0.00192305 0.00013621 0.00192099
5 -154.923753741208 -0.0000003686 0.00034810 0.00002451 0.00068243
6 -154.923753749716 -0.0000000085 0.00017207 0.00001017 0.00014045
7 -154.923753752932 -0.0000000032 0.00001787 0.00000115 0.00005005
8 -154.923753752975 -0.0000000000 0.00000352 0.00000026 0.00000899
9 -154.923753752976 -0.0000000000 0.00000036 0.00000003 0.00000190
* Info * Checkpoint written to file: vlx_20251015_6d4463a8_scf.h5
*** SCF converged in 9 iterations. Time: 0.93 sec.
Spin-Restricted Kohn-Sham:
--------------------------
Total Energy : -154.9237537530 a.u.
Electronic Energy : -236.4626620145 a.u.
Nuclear Repulsion Energy : 81.5389082615 a.u.
------------------------------------
Gradient Norm : 0.0000003617 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
Spin Restricted Orbitals
------------------------
Molecular Orbital No. 9:
--------------------------
Occupation: 2.000 Energy: -0.43037 a.u.
( 1 C 1p0 : -0.34) ( 1 C 2p0 : -0.15) ( 2 C 1p-1: -0.24)
( 2 C 1p0 : 0.17) ( 3 O 1p+1: -0.20) ( 6 H 1s : -0.22)
Molecular Orbital No. 10:
--------------------------
Occupation: 2.000 Energy: -0.38071 a.u.
( 1 C 1p-1: -0.37) ( 1 C 2p-1: -0.16) ( 2 C 1p-1: 0.17)
( 3 O 1p-1: 0.18) ( 4 H 1s : 0.32) ( 6 H 1s : -0.19)
Molecular Orbital No. 11:
--------------------------
Occupation: 2.000 Energy: -0.36918 a.u.
( 1 C 1p+1: 0.31) ( 2 C 1p+1: -0.21) ( 3 O 1p-1: 0.18)
( 3 O 1p0 : 0.25) ( 3 O 2p0 : 0.16) ( 5 H 1s : -0.29)
( 6 H 1s : 0.20) ( 7 H 1s : -0.18)
Molecular Orbital No. 12:
--------------------------
Occupation: 2.000 Energy: -0.32055 a.u.
( 1 C 1p0 : -0.18) ( 2 C 1p+1: -0.16) ( 2 C 1p0 : 0.21)
( 3 O 3s : 0.21) ( 3 O 1p+1: 0.19) ( 3 O 1p-1: -0.15)
( 3 O 1p0 : -0.33) ( 3 O 2p0 : -0.24) ( 7 H 1s : -0.26)
( 7 H 2s : -0.16) ( 8 H 1s : 0.18) ( 9 H 1s : -0.19)
Molecular Orbital No. 13:
--------------------------
Occupation: 2.000 Energy: -0.26783 a.u.
( 1 C 3s : -0.16) ( 1 C 1p0 : 0.17) ( 2 C 1p-1: 0.17)
( 3 O 1p-1: -0.52) ( 3 O 1p0 : 0.22) ( 3 O 2p-1: -0.39)
( 3 O 2p0 : 0.18) ( 8 H 1s : 0.21) ( 8 H 2s : 0.15)
Molecular Orbital No. 14:
--------------------------
Occupation: 0.000 Energy: 0.04880 a.u.
( 1 C 3s : 1.23) ( 1 C 2p+1: -0.29) ( 1 C 2p0 : 0.28)
( 2 C 3s : 0.94) ( 2 C 2p+1: 0.21) ( 2 C 2p-1: 0.28)
( 3 O 3s : 0.52) ( 3 O 1p+1: -0.18) ( 3 O 2p+1: -0.28)
( 4 H 2s : -0.47) ( 5 H 2s : -0.81) ( 6 H 2s : -0.60)
( 7 H 2s : -0.42) ( 8 H 2s : -0.72) ( 9 H 2s : -0.87)
Molecular Orbital No. 15:
--------------------------
Occupation: 0.000 Energy: 0.08807 a.u.
( 1 C 3s : -1.65) ( 1 C 2p+1: -0.16) ( 1 C 2p-1: 0.18)
( 1 C 2p0 : -0.23) ( 2 C 3s : -0.61) ( 2 C 2p+1: -0.39)
( 2 C 2p0 : 0.34) ( 3 O 3s : 0.65) ( 3 O 2p+1: -0.17)
( 4 H 2s : 0.87) ( 5 H 2s : 0.32) ( 6 H 2s : 0.97)
( 7 H 2s : 0.96) ( 8 H 2s : 0.25) ( 9 H 2s : -0.90)
Molecular Orbital No. 16:
--------------------------
Occupation: 0.000 Energy: 0.10591 a.u.
( 1 C 3s : 2.27) ( 1 C 2p+1: -0.28) ( 1 C 2p0 : -0.28)
( 2 C 3s : -2.16) ( 2 C 2p+1: -0.33) ( 2 C 2p0 : -0.44)
( 4 H 2s : -0.92) ( 5 H 2s : -1.29) ( 6 H 2s : -0.16)
( 7 H 2s : 1.19) ( 8 H 2s : 1.10)
Molecular Orbital No. 17:
--------------------------
Occupation: 0.000 Energy: 0.11817 a.u.
( 1 C 1p-1: 0.18) ( 1 C 2p+1: -0.34) ( 1 C 2p-1: 0.45)
( 2 C 3s : 0.55) ( 2 C 1p+1: -0.18) ( 2 C 1p-1: 0.23)
( 2 C 2p+1: -0.44) ( 2 C 2p-1: 0.63) ( 3 O 3s : -0.42)
( 4 H 2s : 0.91) ( 5 H 2s : -0.92) ( 7 H 2s : 0.78)
( 8 H 2s : -1.42) ( 9 H 2s : 0.44)
Molecular Orbital No. 18:
--------------------------
Occupation: 0.000 Energy: 0.13337 a.u.
( 1 C 3s : -0.61) ( 1 C 1p+1: -0.21) ( 1 C 1p-1: -0.16)
( 1 C 1p0 : -0.22) ( 1 C 2p+1: -0.82) ( 1 C 2p-1: -0.60)
( 1 C 2p0 : -0.67) ( 2 C 3s : 1.00) ( 2 C 2p+1: 0.35)
( 4 H 2s : -0.80) ( 5 H 2s : -0.47) ( 6 H 2s : 1.98)
( 7 H 2s : -0.72) ( 8 H 2s : -0.48)
Ground State Dipole Moment
----------------------------
X : -0.288125 a.u. -0.732340 Debye
Y : 0.250781 a.u. 0.637422 Debye
Z : 0.495722 a.u. 1.259998 Debye
Total : 0.625817 a.u. 1.590668 Debye
SCF Gradient Driver
=====================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
================================
Atom Coordinate X Coordinate Y Coordinate Z
C 0.549960827538 0.077025350952 0.837137024260
C 1.088216484782 0.798133082545 -0.390928154863
O 0.391742124693 0.466581889711 -1.575214904917
H 0.560469529523 -1.013887749189 0.683408861715
H -0.491260922071 0.376953159219 1.051766151644
H 1.150798148722 0.310469903933 1.731624025996
H 2.138156433671 0.510470326246 -0.566574684011
H 1.090433262167 1.894504443155 -0.214502591070
H -0.529363054963 0.737227335819 -1.464946249937
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000432999388 0.000404620879 -0.001152290888
C -0.000023650762 -0.000410681166 0.000886488021
O -0.000083503379 -0.000069362202 0.000179296880
H -0.000219429840 -0.000189730494 0.000089360388
H -0.000183109646 -0.000088990261 0.000162774280
H 0.000000143957 0.000121599432 -0.000081952577
H 0.000094827081 0.000141832606 0.000005407903
H 0.000016181687 0.000070730294 -0.000115544390
H -0.000043254883 0.000030137492 0.000014655781
*** Time spent in gradient calculation: 0.90 sec ***
* Info * Energy : -154.9237537530 a.u.
* Info * Gradient : 5.686551e-04 a.u. (RMS)
* Info * 1.295755e-03 a.u. (Max)
* Info * Time : 1.93 sec
* Info * Computing energy and gradient...
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Kohn-Sham
Initial Guess Model : Restart from Checkpoint
Convergence Accelerator : Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Nuclear repulsion energy: 81.5109584362 a.u.
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Molecular grid with 115422 points generated in 0.05 sec.
* Info * Restarting from checkpoint file: vlx_20251015_6d4463a8_scf.h5
* Info * Overlap matrix computed in 0.00 sec.
* Info * Kinetic energy matrix computed in 0.01 sec.
* Info * Nuclear potential matrix computed in 0.00 sec.
* Info * Orthogonalization matrix computed in 0.00 sec.
Iter. | Kohn-Sham Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -154.923093645172 0.0000000000 0.00799309 0.00120147 0.00000000
2 -154.923757127358 -0.0006634822 0.00168248 0.00011018 0.00272612
3 -154.923756988975 0.0000001384 0.00202717 0.00011710 0.00110964
4 -154.923757458054 -0.0000004691 0.00026342 0.00001903 0.00060798
5 -154.923757462937 -0.0000000049 0.00011939 0.00000856 0.00010354
6 -154.923757464345 -0.0000000014 0.00001534 0.00000095 0.00004188
7 -154.923757464372 -0.0000000000 0.00000193 0.00000018 0.00000602
8 -154.923757464372 -0.0000000000 0.00000030 0.00000003 0.00000103
* Info * Checkpoint written to file: vlx_20251015_6d4463a8_scf.h5
*** SCF converged in 8 iterations. Time: 0.80 sec.
Spin-Restricted Kohn-Sham:
--------------------------
Total Energy : -154.9237574644 a.u.
Electronic Energy : -236.4347159006 a.u.
Nuclear Repulsion Energy : 81.5109584362 a.u.
------------------------------------
Gradient Norm : 0.0000002989 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
Spin Restricted Orbitals
------------------------
Molecular Orbital No. 9:
--------------------------
Occupation: 2.000 Energy: -0.43032 a.u.
( 1 C 1p0 : -0.34) ( 1 C 2p0 : -0.15) ( 2 C 1p-1: -0.24)
( 2 C 1p0 : 0.17) ( 3 O 1p+1: -0.20) ( 6 H 1s : -0.22)
Molecular Orbital No. 10:
--------------------------
Occupation: 2.000 Energy: -0.38049 a.u.
( 1 C 1p-1: -0.37) ( 1 C 2p-1: -0.16) ( 2 C 1p-1: 0.17)
( 3 O 1p-1: 0.18) ( 4 H 1s : 0.32) ( 6 H 1s : -0.18)
Molecular Orbital No. 11:
--------------------------
Occupation: 2.000 Energy: -0.36942 a.u.
( 1 C 1p+1: 0.31) ( 2 C 1p+1: -0.21) ( 3 O 1p-1: 0.17)
( 3 O 1p0 : 0.25) ( 3 O 2p0 : 0.16) ( 5 H 1s : -0.29)
( 6 H 1s : 0.20) ( 7 H 1s : -0.18)
Molecular Orbital No. 12:
--------------------------
Occupation: 2.000 Energy: -0.32064 a.u.
( 1 C 1p0 : -0.18) ( 2 C 1p+1: -0.16) ( 2 C 1p0 : 0.21)
( 3 O 3s : 0.21) ( 3 O 1p+1: 0.19) ( 3 O 1p-1: -0.15)
( 3 O 1p0 : -0.33) ( 3 O 2p0 : -0.24) ( 7 H 1s : -0.26)
( 7 H 2s : -0.16) ( 8 H 1s : 0.18) ( 9 H 1s : -0.19)
Molecular Orbital No. 13:
--------------------------
Occupation: 2.000 Energy: -0.26785 a.u.
( 1 C 3s : -0.16) ( 1 C 1p0 : 0.17) ( 2 C 1p-1: 0.17)
( 3 O 1p-1: -0.52) ( 3 O 1p0 : 0.22) ( 3 O 2p-1: -0.39)
( 3 O 2p0 : 0.18) ( 8 H 1s : 0.21) ( 8 H 2s : 0.15)
Molecular Orbital No. 14:
--------------------------
Occupation: 0.000 Energy: 0.04880 a.u.
( 1 C 3s : 1.24) ( 1 C 2p+1: -0.29) ( 1 C 2p0 : 0.28)
( 2 C 3s : 0.94) ( 2 C 2p+1: 0.22) ( 2 C 2p-1: 0.28)
( 3 O 3s : 0.52) ( 3 O 1p+1: -0.18) ( 3 O 2p+1: -0.28)
( 4 H 2s : -0.47) ( 5 H 2s : -0.81) ( 6 H 2s : -0.60)
( 7 H 2s : -0.42) ( 8 H 2s : -0.72) ( 9 H 2s : -0.87)
Molecular Orbital No. 15:
--------------------------
Occupation: 0.000 Energy: 0.08808 a.u.
( 1 C 3s : -1.65) ( 1 C 2p+1: -0.16) ( 1 C 2p-1: 0.18)
( 1 C 2p0 : -0.23) ( 2 C 3s : -0.61) ( 2 C 2p+1: -0.39)
( 2 C 2p0 : 0.34) ( 3 O 3s : 0.65) ( 3 O 2p+1: -0.17)
( 4 H 2s : 0.87) ( 5 H 2s : 0.32) ( 6 H 2s : 0.97)
( 7 H 2s : 0.96) ( 8 H 2s : 0.25) ( 9 H 2s : -0.90)
Molecular Orbital No. 16:
--------------------------
Occupation: 0.000 Energy: 0.10607 a.u.
( 1 C 3s : 2.27) ( 1 C 2p+1: -0.28) ( 1 C 2p0 : -0.28)
( 2 C 3s : -2.17) ( 2 C 2p+1: -0.33) ( 2 C 2p0 : -0.45)
( 4 H 2s : -0.92) ( 5 H 2s : -1.29) ( 6 H 2s : -0.16)
( 7 H 2s : 1.19) ( 8 H 2s : 1.10)
Molecular Orbital No. 17:
--------------------------
Occupation: 0.000 Energy: 0.11827 a.u.
( 1 C 1p-1: 0.18) ( 1 C 2p+1: -0.35) ( 1 C 2p-1: 0.44)
( 2 C 3s : 0.55) ( 2 C 1p+1: -0.18) ( 2 C 1p-1: 0.23)
( 2 C 2p+1: -0.44) ( 2 C 2p-1: 0.63) ( 3 O 3s : -0.42)
( 4 H 2s : 0.91) ( 5 H 2s : -0.93) ( 7 H 2s : 0.79)
( 8 H 2s : -1.42) ( 9 H 2s : 0.44)
Molecular Orbital No. 18:
--------------------------
Occupation: 0.000 Energy: 0.13329 a.u.
( 1 C 3s : -0.61) ( 1 C 1p+1: -0.21) ( 1 C 1p-1: -0.16)
( 1 C 1p0 : -0.22) ( 1 C 2p+1: -0.82) ( 1 C 2p-1: -0.60)
( 1 C 2p0 : -0.67) ( 2 C 3s : 0.99) ( 2 C 2p+1: 0.35)
( 4 H 2s : -0.81) ( 5 H 2s : -0.47) ( 6 H 2s : 1.98)
( 7 H 2s : -0.72) ( 8 H 2s : -0.48)
Ground State Dipole Moment
----------------------------
X : -0.288891 a.u. -0.734288 Debye
Y : 0.248896 a.u. 0.632630 Debye
Z : 0.495830 a.u. 1.260274 Debye
Total : 0.625503 a.u. 1.589871 Debye
SCF Gradient Driver
=====================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
================================
Atom Coordinate X Coordinate Y Coordinate Z
C 0.549817038982 0.076846210000 0.838536601829
C 1.088064856050 0.798462485633 -0.391391704536
O 0.392277969783 0.467713906316 -1.576228965933
H 0.561598913238 -1.013580778330 0.683257123441
H -0.491097563374 0.377477485974 1.052141581273
H 1.150830830109 0.309296648834 1.733170868343
H 2.137570958533 0.509304673925 -0.566686358452
H 1.090771690813 1.894541943074 -0.213830152308
H -0.529457925340 0.735980843860 -1.465538315022
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000089409870 0.000052756735 -0.000147431360
C -0.000024796877 -0.000058188210 0.000144822971
O -0.000010261115 -0.000010731995 -0.000065297828
H -0.000037747893 -0.000015584259 0.000008891924
H -0.000020414553 0.000005641881 0.000031166027
H -0.000008842773 0.000001798525 0.000017963618
H 0.000024232119 0.000025699654 0.000004934284
H -0.000005270692 0.000008131698 -0.000024763568
H -0.000015080522 0.000000689201 0.000018106418
*** Time spent in gradient calculation: 1.04 sec ***
* Info * Energy : -154.9237574644 a.u.
* Info * Gradient : 8.697246e-05 a.u. (RMS)
* Info * 1.803147e-04 a.u. (Max)
* Info * Time : 1.93 sec
* Info * Computing energy and gradient...
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Kohn-Sham
Initial Guess Model : Restart from Checkpoint
Convergence Accelerator : Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Nuclear repulsion energy: 81.5085263241 a.u.
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Molecular grid with 115422 points generated in 0.06 sec.
* Info * Restarting from checkpoint file: vlx_20251015_6d4463a8_scf.h5
* Info * Overlap matrix computed in 0.00 sec.
* Info * Kinetic energy matrix computed in 0.01 sec.
* Info * Nuclear potential matrix computed in 0.00 sec.
* Info * Orthogonalization matrix computed in 0.00 sec.
Iter. | Kohn-Sham Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -154.923718837739 0.0000000000 0.00200793 0.00020250 0.00000000
2 -154.923757571185 -0.0000387334 0.00051349 0.00005208 0.00115036
3 -154.923757578046 -0.0000000069 0.00041269 0.00002341 0.00026320
4 -154.923757593538 -0.0000000155 0.00016581 0.00001071 0.00015350
5 -154.923757596218 -0.0000000027 0.00002459 0.00000158 0.00004844
6 -154.923757596265 -0.0000000000 0.00001143 0.00000063 0.00001101
7 -154.923757596280 -0.0000000000 0.00000065 0.00000004 0.00000389
* Info * Checkpoint written to file: vlx_20251015_6d4463a8_scf.h5
*** SCF converged in 7 iterations. Time: 0.92 sec.
Spin-Restricted Kohn-Sham:
--------------------------
Total Energy : -154.9237575963 a.u.
Electronic Energy : -236.4322839204 a.u.
Nuclear Repulsion Energy : 81.5085263241 a.u.
------------------------------------
Gradient Norm : 0.0000006509 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
Spin Restricted Orbitals
------------------------
Molecular Orbital No. 9:
--------------------------
Occupation: 2.000 Energy: -0.43024 a.u.
( 1 C 1p0 : -0.34) ( 1 C 2p0 : -0.15) ( 2 C 1p-1: -0.24)
( 2 C 1p0 : 0.17) ( 3 O 1p+1: -0.20) ( 6 H 1s : -0.22)
Molecular Orbital No. 10:
--------------------------
Occupation: 2.000 Energy: -0.38047 a.u.
( 1 C 1p-1: -0.37) ( 1 C 2p-1: -0.16) ( 2 C 1p-1: 0.17)
( 3 O 1p-1: 0.18) ( 4 H 1s : 0.32) ( 6 H 1s : -0.18)
Molecular Orbital No. 11:
--------------------------
Occupation: 2.000 Energy: -0.36945 a.u.
( 1 C 1p+1: 0.31) ( 2 C 1p+1: -0.21) ( 3 O 1p-1: 0.17)
( 3 O 1p0 : 0.25) ( 3 O 2p0 : 0.16) ( 5 H 1s : -0.29)
( 6 H 1s : 0.20) ( 7 H 1s : -0.18)
Molecular Orbital No. 12:
--------------------------
Occupation: 2.000 Energy: -0.32062 a.u.
( 1 C 1p0 : -0.18) ( 2 C 1p+1: -0.16) ( 2 C 1p0 : 0.21)
( 3 O 3s : 0.21) ( 3 O 1p+1: 0.19) ( 3 O 1p-1: -0.15)
( 3 O 1p0 : -0.33) ( 3 O 2p0 : -0.24) ( 7 H 1s : -0.26)
( 7 H 2s : -0.16) ( 8 H 1s : 0.18) ( 9 H 1s : -0.19)
Molecular Orbital No. 13:
--------------------------
Occupation: 2.000 Energy: -0.26785 a.u.
( 1 C 3s : -0.16) ( 1 C 1p0 : 0.17) ( 2 C 1p-1: 0.17)
( 3 O 1p-1: -0.52) ( 3 O 1p0 : 0.22) ( 3 O 2p-1: -0.39)
( 3 O 2p0 : 0.18) ( 8 H 1s : 0.21) ( 8 H 2s : 0.15)
Molecular Orbital No. 14:
--------------------------
Occupation: 0.000 Energy: 0.04880 a.u.
( 1 C 3s : 1.23) ( 1 C 2p+1: -0.29) ( 1 C 2p0 : 0.28)
( 2 C 3s : 0.94) ( 2 C 2p+1: 0.22) ( 2 C 2p-1: 0.28)
( 3 O 3s : 0.52) ( 3 O 1p+1: -0.18) ( 3 O 2p+1: -0.28)
( 4 H 2s : -0.47) ( 5 H 2s : -0.81) ( 6 H 2s : -0.60)
( 7 H 2s : -0.42) ( 8 H 2s : -0.72) ( 9 H 2s : -0.87)
Molecular Orbital No. 15:
--------------------------
Occupation: 0.000 Energy: 0.08807 a.u.
( 1 C 3s : -1.65) ( 1 C 2p+1: -0.15) ( 1 C 2p-1: 0.18)
( 1 C 2p0 : -0.23) ( 2 C 3s : -0.61) ( 2 C 2p+1: -0.39)
( 2 C 2p0 : 0.34) ( 3 O 3s : 0.65) ( 3 O 2p+1: -0.17)
( 4 H 2s : 0.87) ( 5 H 2s : 0.32) ( 6 H 2s : 0.97)
( 7 H 2s : 0.96) ( 8 H 2s : 0.25) ( 9 H 2s : -0.90)
Molecular Orbital No. 16:
--------------------------
Occupation: 0.000 Energy: 0.10610 a.u.
( 1 C 3s : 2.27) ( 1 C 2p+1: -0.28) ( 1 C 2p0 : -0.28)
( 2 C 3s : -2.17) ( 2 C 2p+1: -0.33) ( 2 C 2p0 : -0.45)
( 4 H 2s : -0.91) ( 5 H 2s : -1.29) ( 6 H 2s : -0.16)
( 7 H 2s : 1.19) ( 8 H 2s : 1.10)
Molecular Orbital No. 17:
--------------------------
Occupation: 0.000 Energy: 0.11831 a.u.
( 1 C 1p-1: 0.18) ( 1 C 2p+1: -0.35) ( 1 C 2p-1: 0.44)
( 2 C 3s : 0.55) ( 2 C 1p+1: -0.18) ( 2 C 1p-1: 0.23)
( 2 C 2p+1: -0.44) ( 2 C 2p-1: 0.63) ( 3 O 3s : -0.42)
( 4 H 2s : 0.91) ( 5 H 2s : -0.93) ( 7 H 2s : 0.79)
( 8 H 2s : -1.42) ( 9 H 2s : 0.44)
Molecular Orbital No. 18:
--------------------------
Occupation: 0.000 Energy: 0.13331 a.u.
( 1 C 3s : -0.61) ( 1 C 1p+1: -0.21) ( 1 C 1p-1: -0.16)
( 1 C 1p0 : -0.22) ( 1 C 2p+1: -0.82) ( 1 C 2p-1: -0.60)
( 1 C 2p0 : -0.67) ( 2 C 3s : 0.99) ( 2 C 2p+1: 0.35)
( 4 H 2s : -0.81) ( 5 H 2s : -0.47) ( 6 H 2s : 1.98)
( 7 H 2s : -0.72) ( 8 H 2s : -0.48)
Ground State Dipole Moment
----------------------------
X : -0.288803 a.u. -0.734065 Debye
Y : 0.249793 a.u. 0.634911 Debye
Z : 0.495071 a.u. 1.258346 Debye
Total : 0.625220 a.u. 1.589150 Debye
SCF Gradient Driver
=====================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
================================
Atom Coordinate X Coordinate Y Coordinate Z
C 0.549869085078 0.076603678990 0.838953188304
C 1.088032860166 0.798258678384 -0.391293917872
O 0.392245013578 0.467397567247 -1.576018184414
H 0.562296299504 -1.013777738821 0.683630680225
H -0.491128475009 0.376952161094 1.052254598319
H 1.150916061074 0.309365651428 1.733464090295
H 2.137448338388 0.508874233506 -0.566586126234
H 1.090780186443 1.894308542885 -0.213646953467
H -0.529184234753 0.736988415446 -1.466103751380
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000007830985 0.000000593366 -0.000002966531
C 0.000006496757 0.000001093137 0.000017775692
O -0.000002175899 -0.000011997122 -0.000031445230
H -0.000008469445 0.000004757840 -0.000004725335
H 0.000002921984 0.000011196930 0.000006831042
H 0.000000217716 -0.000007451809 0.000005648227
H 0.000002958924 0.000009284251 0.000002974971
H -0.000010383743 0.000000431559 -0.000006320969
H -0.000008046952 0.000002336829 0.000000573716
*** Time spent in gradient calculation: 1.04 sec ***
* Info * Energy : -154.9237575963 a.u.
* Info * Gradient : 1.589682e-05 a.u. (RMS)
* Info * 3.372637e-05 a.u. (Max)
* Info * Time : 2.05 sec
* Info * Geometry optimization completed.
Molecular Geometry (Angstroms)
================================
Atom Coordinate X Coordinate Y Coordinate Z
C 0.549869085078 0.076603678990 0.838953188304
C 1.088032860166 0.798258678384 -0.391293917872
O 0.392245013578 0.467397567247 -1.576018184414
H 0.562296299504 -1.013777738821 0.683630680225
H -0.491128475009 0.376952161094 1.052254598319
H 1.150916061074 0.309365651428 1.733464090295
H 2.137448338388 0.508874233506 -0.566586126234
H 1.090780186443 1.894308542885 -0.213646953467
H -0.529184234753 0.736988415446 -1.466103751380
Summary of Geometry Optimization
==================================
Opt.Step Energy (a.u.) Energy Change (a.u.) Displacement (RMS, Max)
-------------------------------------------------------------------------------------
0 -154.921531860738 0.000000000000 0.000e+00 0.000e+00
1 -154.923412038861 -0.001880178122 4.163e-02 7.222e-02
2 -154.923535125738 -0.000123086877 1.222e-02 2.812e-02
3 -154.923634203799 -0.000099078061 1.734e-02 4.277e-02
4 -154.923730200268 -0.000095996470 2.705e-02 6.652e-02
5 -154.923753752976 -0.000023552708 8.954e-03 2.128e-02
6 -154.923757464372 -0.000003711396 1.246e-03 1.723e-03
7 -154.923757596280 -0.000000131908 5.537e-04 1.250e-03
Statistical Deviation between
Optimized Geometry and Initial Geometry
=========================================
Internal Coord. RMS deviation Max. deviation
-----------------------------------------------------------
Bonds 0.012 Angstrom 0.027 Angstrom
Angles 1.504 degree 2.870 degree
Dihedrals 7.997 degree 17.617 degree
*** Time spent in Optimization Driver: 16.78 sec
opt_drv.show_convergence(opt_results)Loading...
From an h5 file
opt_results = vlx.read_results("../output_files/bithio-S0-opt.h5", label="opt")
opt_drv.show_convergence(opt_results)Loading...
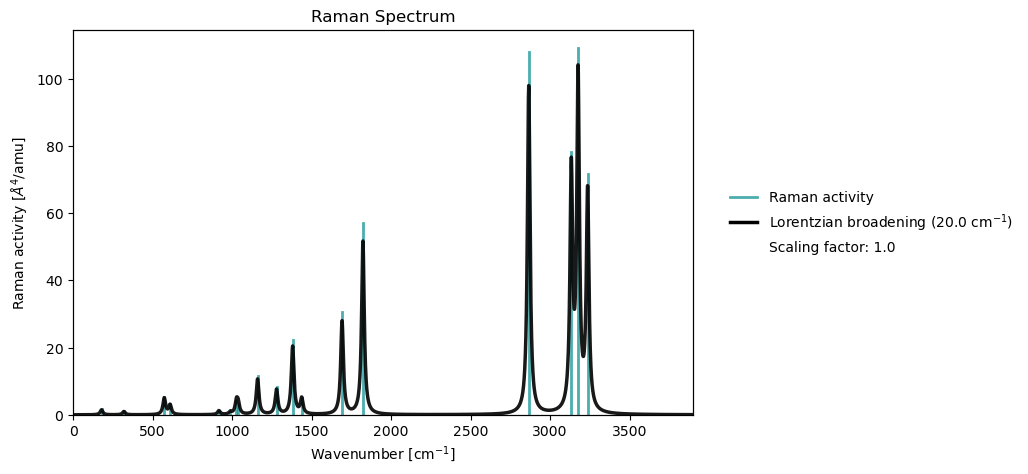
Spectrum plotting¶
From a notebook cell
molecule = vlx.Molecule.read_smiles('CCO')
basis = vlx.MolecularBasis.read(molecule, 'def2-svp')
scf_drv = vlx.ScfRestrictedDriver()
scf_drv.xcfun = 'b3lyp'
scf_results = scf_drv.compute(molecule, basis)
rsp_drv = vlx.lreigensolver.LinearResponseEigenSolver()
rsp_drv.nstates = 10
rsp_results = rsp_drv.compute(molecule, basis, scf_results)
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Kohn-Sham
Initial Guess Model : Superposition of Atomic Densities
Convergence Accelerator : Two Level Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Nuclear repulsion energy: 81.6235421941 a.u.
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Molecular grid with 115780 points generated in 0.04 sec.
* Info * Overlap matrix computed in 0.00 sec.
* Info * Kinetic energy matrix computed in 0.00 sec.
* Info * Nuclear potential matrix computed in 0.00 sec.
* Info * Orthogonalization matrix computed in 0.00 sec.
* Info * Starting Reduced Basis SCF calculation...
* Info * ...done. SCF energy in reduced basis set: -153.879535889192 a.u. Time: 0.09 sec.
* Info * Overlap matrix computed in 0.00 sec.
* Info * Kinetic energy matrix computed in 0.00 sec.
* Info * Nuclear potential matrix computed in 0.00 sec.
* Info * Orthogonalization matrix computed in 0.00 sec.
Iter. | Kohn-Sham Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -154.914794089345 0.0000000000 0.25018147 0.01667989 0.00000000
2 -154.917119603355 -0.0023255140 0.19462265 0.01472423 0.14964060
3 -154.921762137858 -0.0046425345 0.03453589 0.00229114 0.06939501
4 -154.921897198299 -0.0001350604 0.00585503 0.00048445 0.01097431
5 -154.921899439895 -0.0000022416 0.00321331 0.00025903 0.00264140
6 -154.921900382713 -0.0000009428 0.00024575 0.00001516 0.00096058
7 -154.921900388972 -0.0000000063 0.00007656 0.00000428 0.00009534
8 -154.921900389669 -0.0000000007 0.00001457 0.00000103 0.00002510
9 -154.921900389694 -0.0000000000 0.00000174 0.00000014 0.00000525
10 -154.921900389695 -0.0000000000 0.00000022 0.00000002 0.00000085
*** SCF converged in 10 iterations. Time: 1.13 sec.
Spin-Restricted Kohn-Sham:
--------------------------
Total Energy : -154.9219003897 a.u.
Electronic Energy : -236.5454425838 a.u.
Nuclear Repulsion Energy : 81.6235421941 a.u.
------------------------------------
Gradient Norm : 0.0000002248 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
Spin Restricted Orbitals
------------------------
Molecular Orbital No. 9:
--------------------------
Occupation: 2.000 Energy: -0.40059 a.u.
( 1 C 1p0 : 0.27) ( 2 C 1p+1: 0.20) ( 2 C 1p0 : -0.26)
( 3 O 3s : -0.18) ( 3 O 1p-1: -0.34) ( 3 O 2p-1: -0.20)
Molecular Orbital No. 10:
--------------------------
Occupation: 2.000 Energy: -0.37987 a.u.
( 1 C 1p-1: -0.39) ( 6 H 1s : 0.38) ( 6 H 2s : 0.19)
Molecular Orbital No. 11:
--------------------------
Occupation: 2.000 Energy: -0.36361 a.u.
( 1 C 1p+1: -0.31) ( 1 C 1p0 : -0.19) ( 2 C 1p+1: 0.15)
( 3 O 1p+1: 0.18) ( 4 H 1s : -0.28) ( 5 H 1s : 0.28)
Molecular Orbital No. 12:
--------------------------
Occupation: 2.000 Energy: -0.32392 a.u.
( 1 C 3s : 0.16) ( 1 C 1p0 : -0.24) ( 2 C 1p-1: 0.19)
( 2 C 1p0 : 0.23) ( 3 O 3s : -0.24) ( 3 O 1p-1: -0.31)
( 3 O 1p0 : -0.27) ( 3 O 2p-1: -0.21) ( 3 O 2p0 : -0.17)
( 7 H 1s : -0.16) ( 8 H 1s : -0.16) ( 9 H 1s : 0.20)
Molecular Orbital No. 13:
--------------------------
Occupation: 2.000 Energy: -0.26363 a.u.
( 2 C 1p+1: -0.16) ( 3 O 1p+1: 0.47) ( 3 O 1p0 : 0.29)
( 3 O 2p+1: 0.36) ( 3 O 2p0 : 0.22) ( 7 H 1s : -0.23)
( 7 H 2s : -0.17) ( 8 H 1s : 0.23) ( 8 H 2s : 0.17)
Molecular Orbital No. 14:
--------------------------
Occupation: 0.000 Energy: 0.04506 a.u.
( 1 C 3s : 0.50) ( 1 C 2p+1: -0.16) ( 1 C 2p0 : 0.21)
( 2 C 3s : 1.29) ( 2 C 1p-1: -0.18) ( 2 C 2p-1: -0.34)
( 2 C 2p0 : -0.17) ( 3 O 3s : 0.55) ( 3 O 1p0 : -0.17)
( 3 O 2p+1: 0.16) ( 3 O 2p0 : -0.25) ( 4 H 2s : -0.22)
( 5 H 2s : -0.22) ( 6 H 2s : -0.51) ( 7 H 2s : -0.73)
( 8 H 2s : -0.73) ( 9 H 2s : -1.00)
Molecular Orbital No. 15:
--------------------------
Occupation: 0.000 Energy: 0.08189 a.u.
( 1 C 2s : 0.16) ( 1 C 3s : 2.26) ( 1 C 2p0 : 0.24)
( 2 C 3s : 0.31) ( 2 C 2p+1: 0.20) ( 2 C 2p0 : -0.36)
( 3 O 3s : -0.44) ( 3 O 1p0 : 0.16) ( 3 O 2p0 : 0.23)
( 4 H 2s : -0.98) ( 5 H 2s : -0.98) ( 6 H 2s : -0.92)
( 7 H 2s : -0.44) ( 8 H 2s : -0.44) ( 9 H 2s : 0.68)
Molecular Orbital No. 16:
--------------------------
Occupation: 0.000 Energy: 0.11463 a.u.
( 1 C 1p+1: 0.19) ( 1 C 2p+1: 0.48) ( 1 C 2p0 : 0.30)
( 2 C 1p+1: 0.27) ( 2 C 1p0 : 0.17) ( 2 C 2p+1: 0.72)
( 2 C 2p-1: -0.22) ( 2 C 2p0 : 0.45) ( 4 H 2s : -0.92)
( 5 H 2s : 0.92) ( 7 H 2s : -1.25) ( 8 H 2s : 1.25)
Molecular Orbital No. 17:
--------------------------
Occupation: 0.000 Energy: 0.12054 a.u.
( 1 C 3s : -2.12) ( 1 C 2p-1: -0.17) ( 2 C 2s : 0.18)
( 2 C 3s : 2.39) ( 2 C 2p-1: -0.37) ( 3 O 3s : -0.45)
( 4 H 2s : 0.89) ( 5 H 2s : 0.89) ( 6 H 2s : 0.39)
( 7 H 2s : -1.27) ( 8 H 2s : -1.27) ( 9 H 2s : 0.58)
Molecular Orbital No. 18:
--------------------------
Occupation: 0.000 Energy: 0.13626 a.u.
( 1 C 3s : -0.33) ( 1 C 1p-1: 0.36) ( 1 C 2p+1: 0.26)
( 1 C 2p-1: 1.25) ( 1 C 2p0 : 0.20) ( 2 C 3s : 0.54)
( 2 C 2p-1: -0.35) ( 4 H 2s : -0.77) ( 5 H 2s : -0.77)
( 6 H 2s : 1.98) ( 7 H 2s : -0.38) ( 8 H 2s : -0.38)
Ground State Dipole Moment
----------------------------
X : 0.125867 a.u. 0.319921 Debye
Y : -0.428651 a.u. -1.089523 Debye
Z : -0.411141 a.u. -1.045017 Debye
Total : 0.607142 a.u. 1.543201 Debye
Linear Response EigenSolver Setup
===================================
Number of States : 10
Max. Number of Iterations : 150
Convergence Threshold : 1.0e-04
ERI Screening Threshold : 1.0e-12
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Molecular grid with 115780 points generated in 0.04 sec.
* Info * Processing 10 Fock builds...
* Info * 10 gerade trial vectors in reduced space
* Info * 10 ungerade trial vectors in reduced space
* Info * 369.30 kB of memory used for subspace procedure on the master node
* Info * 4.72 GB of memory available for the solver on the master node
*** Iteration: 1 * Residuals (Max,Min): 3.41e-01 and 4.50e-02
Excitation 1 : 0.25828353 Residual Norm: 0.12975868
Excitation 2 : 0.30512048 Residual Norm: 0.07988991
Excitation 3 : 0.32853298 Residual Norm: 0.18878606
Excitation 4 : 0.34455950 Residual Norm: 0.14812874
Excitation 5 : 0.34740410 Residual Norm: 0.08704518
Excitation 6 : 0.36448401 Residual Norm: 0.04499622
Excitation 7 : 0.37107383 Residual Norm: 0.05970524
Excitation 8 : 0.37217510 Residual Norm: 0.14530301
Excitation 9 : 0.39477281 Residual Norm: 0.11598177
Excitation 10 : 0.40627722 Residual Norm: 0.34144002
* Info * Processing 10 Fock builds...
* Info * 20 gerade trial vectors in reduced space
* Info * 20 ungerade trial vectors in reduced space
* Info * 614.74 kB of memory used for subspace procedure on the master node
* Info * 4.71 GB of memory available for the solver on the master node
*** Iteration: 2 * Residuals (Max,Min): 5.12e-02 and 9.31e-03
Excitation 1 : 0.25643695 Residual Norm: 0.01484580
Excitation 2 : 0.30412481 Residual Norm: 0.00931293
Excitation 3 : 0.32458225 Residual Norm: 0.05120032
Excitation 4 : 0.34077197 Residual Norm: 0.04130548
Excitation 5 : 0.34595515 Residual Norm: 0.01716263
Excitation 6 : 0.36392693 Residual Norm: 0.01321176
Excitation 7 : 0.36866623 Residual Norm: 0.04043321
Excitation 8 : 0.36986822 Residual Norm: 0.02229708
Excitation 9 : 0.39146749 Residual Norm: 0.03014471
Excitation 10 : 0.39217355 Residual Norm: 0.04394602
* Info * Processing 10 Fock builds...
* Info * 30 gerade trial vectors in reduced space
* Info * 30 ungerade trial vectors in reduced space
* Info * 860.18 kB of memory used for subspace procedure on the master node
* Info * 4.71 GB of memory available for the solver on the master node
*** Iteration: 3 * Residuals (Max,Min): 1.93e-02 and 1.23e-03
Excitation 1 : 0.25637448 Residual Norm: 0.00152584
Excitation 2 : 0.30409700 Residual Norm: 0.00123134
Excitation 3 : 0.32397725 Residual Norm: 0.01222466
Excitation 4 : 0.34042901 Residual Norm: 0.00907053
Excitation 5 : 0.34587013 Residual Norm: 0.00433759
Excitation 6 : 0.36385388 Residual Norm: 0.00458902
Excitation 7 : 0.36812301 Residual Norm: 0.01675624
Excitation 8 : 0.36973132 Residual Norm: 0.00522631
Excitation 9 : 0.39087642 Residual Norm: 0.01930215
Excitation 10 : 0.39164530 Residual Norm: 0.01265875
* Info * Processing 10 Fock builds...
* Info * 40 gerade trial vectors in reduced space
* Info * 40 ungerade trial vectors in reduced space
* Info * 1.11 MB of memory used for subspace procedure on the master node
* Info * 4.69 GB of memory available for the solver on the master node
*** Iteration: 4 * Residuals (Max,Min): 9.74e-03 and 1.68e-04
Excitation 1 : 0.25637392 Residual Norm: 0.00016779
Excitation 2 : 0.30409654 Residual Norm: 0.00018704
Excitation 3 : 0.32393942 Residual Norm: 0.00251031
Excitation 4 : 0.34040964 Residual Norm: 0.00182205
Excitation 5 : 0.34586213 Residual Norm: 0.00091325
Excitation 6 : 0.36384334 Residual Norm: 0.00116032
Excitation 7 : 0.36802739 Residual Norm: 0.00615261
Excitation 8 : 0.36971404 Residual Norm: 0.00159314
Excitation 9 : 0.39071597 Residual Norm: 0.00973928
Excitation 10 : 0.39156417 Residual Norm: 0.00778579
* Info * Processing 10 Fock builds...
* Info * 50 gerade trial vectors in reduced space
* Info * 50 ungerade trial vectors in reduced space
* Info * 1.35 MB of memory used for subspace procedure on the master node
* Info * 4.69 GB of memory available for the solver on the master node
*** Iteration: 5 * Residuals (Max,Min): 3.50e-03 and 1.22e-05
Excitation 1 : 0.25637392 Residual Norm: 0.00001220 converged
Excitation 2 : 0.30409653 Residual Norm: 0.00001400 converged
Excitation 3 : 0.32393813 Residual Norm: 0.00032400
Excitation 4 : 0.34040892 Residual Norm: 0.00028153
Excitation 5 : 0.34586184 Residual Norm: 0.00014343
Excitation 6 : 0.36384283 Residual Norm: 0.00021891
Excitation 7 : 0.36801464 Residual Norm: 0.00143175
Excitation 8 : 0.36971286 Residual Norm: 0.00034982
Excitation 9 : 0.39066726 Residual Norm: 0.00350152
Excitation 10 : 0.39153322 Residual Norm: 0.00264597
* Info * Processing 8 Fock builds...
* Info * 58 gerade trial vectors in reduced space
* Info * 58 ungerade trial vectors in reduced space
* Info * 1.55 MB of memory used for subspace procedure on the master node
* Info * 4.70 GB of memory available for the solver on the master node
*** Iteration: 6 * Residuals (Max,Min): 8.88e-04 and 6.14e-06
Excitation 1 : 0.25637392 Residual Norm: 0.00001031 converged
Excitation 2 : 0.30409653 Residual Norm: 0.00000614 converged
Excitation 3 : 0.32393810 Residual Norm: 0.00003231 converged
Excitation 4 : 0.34040890 Residual Norm: 0.00003755 converged
Excitation 5 : 0.34586183 Residual Norm: 0.00001354 converged
Excitation 6 : 0.36384282 Residual Norm: 0.00002026 converged
Excitation 7 : 0.36801397 Residual Norm: 0.00025463
Excitation 8 : 0.36971283 Residual Norm: 0.00003570 converged
Excitation 9 : 0.39066207 Residual Norm: 0.00088850
Excitation 10 : 0.39153101 Residual Norm: 0.00055455
* Info * Processing 3 Fock builds...
* Info * 61 gerade trial vectors in reduced space
* Info * 61 ungerade trial vectors in reduced space
* Info * 1.62 MB of memory used for subspace procedure on the master node
* Info * 4.64 GB of memory available for the solver on the master node
*** Iteration: 7 * Residuals (Max,Min): 1.43e-04 and 6.14e-06
Excitation 1 : 0.25637392 Residual Norm: 0.00001031 converged
Excitation 2 : 0.30409653 Residual Norm: 0.00000614 converged
Excitation 3 : 0.32393810 Residual Norm: 0.00002531 converged
Excitation 4 : 0.34040890 Residual Norm: 0.00001768 converged
Excitation 5 : 0.34586183 Residual Norm: 0.00001354 converged
Excitation 6 : 0.36384282 Residual Norm: 0.00002026 converged
Excitation 7 : 0.36801395 Residual Norm: 0.00003074 converged
Excitation 8 : 0.36971283 Residual Norm: 0.00003570 converged
Excitation 9 : 0.39066186 Residual Norm: 0.00014267
Excitation 10 : 0.39153094 Residual Norm: 0.00007834 converged
* Info * Processing 1 Fock build...
* Info * 62 gerade trial vectors in reduced space
* Info * 62 ungerade trial vectors in reduced space
* Info * 1.65 MB of memory used for subspace procedure on the master node
* Info * 4.64 GB of memory available for the solver on the master node
*** Iteration: 8 * Residuals (Max,Min): 6.50e-05 and 6.14e-06
Excitation 1 : 0.25637392 Residual Norm: 0.00001031 converged
Excitation 2 : 0.30409653 Residual Norm: 0.00000614 converged
Excitation 3 : 0.32393810 Residual Norm: 0.00002473 converged
Excitation 4 : 0.34040890 Residual Norm: 0.00001763 converged
Excitation 5 : 0.34586183 Residual Norm: 0.00001354 converged
Excitation 6 : 0.36384282 Residual Norm: 0.00002026 converged
Excitation 7 : 0.36801395 Residual Norm: 0.00001792 converged
Excitation 8 : 0.36971283 Residual Norm: 0.00003570 converged
Excitation 9 : 0.39066185 Residual Norm: 0.00002608 converged
Excitation 10 : 0.39153094 Residual Norm: 0.00006497 converged
*** Linear response converged in 8 iterations. Time: 5.44 sec
Electric Transition Dipole Moments (dipole length, a.u.)
--------------------------------------------------------
X Y Z
Excited State S1: -0.096584 0.029362 -0.060178
Excited State S2: 0.156821 -0.047672 0.097714
Excited State S3: -0.114830 -0.113544 0.128900
Excited State S4: -0.079130 0.456403 0.349650
Excited State S5: -0.468361 0.142376 -0.291828
Excited State S6: 0.020215 -0.006143 0.012598
Excited State S7: -0.060463 -0.158777 0.019570
Excited State S8: 0.053389 -0.016234 0.033268
Excited State S9: -0.320639 -0.556426 0.243135
Excited State S10: -0.185092 -0.232673 0.183534
Electric Transition Dipole Moments (dipole velocity, a.u.)
----------------------------------------------------------
X Y Z
Excited State S1: 0.087674 -0.026650 0.054628
Excited State S2: 0.269372 -0.081885 0.167842
Excited State S3: -0.145545 -0.037589 0.215256
Excited State S4: -0.017195 0.531369 0.286819
Excited State S5: -0.530540 0.161278 -0.330568
Excited State S6: 0.013768 -0.004183 0.008580
Excited State S7: -0.029359 -0.166109 -0.033930
Excited State S8: 0.078880 -0.023983 0.049150
Excited State S9: -0.325020 -0.605501 0.226224
Excited State S10: -0.153227 -0.271852 0.113275
Magnetic Transition Dipole Moments (a.u.)
-----------------------------------------
X Y Z
Excited State S1: 0.141941 -0.132449 -0.292428
Excited State S2: -0.032742 -0.189124 -0.039728
Excited State S3: 0.426759 -0.168188 0.259187
Excited State S4: -0.249194 0.021898 -0.055502
Excited State S5: -0.125575 -0.159279 0.123830
Excited State S6: 0.116703 0.007847 -0.183474
Excited State S7: -0.198882 0.067347 -0.157635
Excited State S8: -0.086085 -0.311772 -0.013952
Excited State S9: 0.742352 -0.274406 0.332083
Excited State S10: 0.057804 -0.042036 -0.022686
One-Photon Absorption
---------------------
Excited State S1: 0.25637392 a.u. 6.97629 eV Osc.Str. 0.0024
Excited State S2: 0.30409653 a.u. 8.27489 eV Osc.Str. 0.0074
Excited State S3: 0.32393810 a.u. 8.81480 eV Osc.Str. 0.0092
Excited State S4: 0.34040890 a.u. 9.26300 eV Osc.Str. 0.0764
Excited State S5: 0.34586183 a.u. 9.41138 eV Osc.Str. 0.0749
Excited State S6: 0.36384282 a.u. 9.90067 eV Osc.Str. 0.0001
Excited State S7: 0.36801395 a.u. 10.01417 eV Osc.Str. 0.0072
Excited State S8: 0.36971283 a.u. 10.06040 eV Osc.Str. 0.0010
Excited State S9: 0.39066185 a.u. 10.63045 eV Osc.Str. 0.1228
Excited State S10: 0.39153094 a.u. 10.65410 eV Osc.Str. 0.0319
Electronic Circular Dichroism
-----------------------------
Excited State S1: Rot.Str. -0.000001 a.u. -0.0003 [10**(-40) cgs]
Excited State S2: Rot.Str. -0.000001 a.u. -0.0006 [10**(-40) cgs]
Excited State S3: Rot.Str. 0.000001 a.u. 0.0004 [10**(-40) cgs]
Excited State S4: Rot.Str. 0.000002 a.u. 0.0008 [10**(-40) cgs]
Excited State S5: Rot.Str. 0.000000 a.u. 0.0001 [10**(-40) cgs]
Excited State S6: Rot.Str. -0.000000 a.u. -0.0001 [10**(-40) cgs]
Excited State S7: Rot.Str. 0.000001 a.u. 0.0002 [10**(-40) cgs]
Excited State S8: Rot.Str. 0.000001 a.u. 0.0006 [10**(-40) cgs]
Excited State S9: Rot.Str. -0.000001 a.u. -0.0004 [10**(-40) cgs]
Excited State S10: Rot.Str. 0.000001 a.u. 0.0004 [10**(-40) cgs]
Character of excitations:
Excited state 1
---------------
HOMO -> LUMO 0.9830
Excited state 2
---------------
HOMO -> LUMO+1 -0.9756
Excited state 3
---------------
HOMO-1 -> LUMO -0.9902
Excited state 4
---------------
HOMO -> LUMO+2 0.9882
Excited state 5
---------------
HOMO -> LUMO+3 0.9882
Excited state 6
---------------
HOMO -> LUMO+4 0.9711
HOMO-2 -> LUMO 0.2196
Excited state 7
---------------
HOMO-1 -> LUMO+1 -0.9433
HOMO -> LUMO+5 -0.2606
Excited state 8
---------------
HOMO-2 -> LUMO 0.9647
HOMO -> LUMO+4 -0.2260
Excited state 9
---------------
HOMO-3 -> LUMO 0.9551
HOMO-4 -> LUMO 0.2360
Excited state 10
----------------
HOMO -> LUMO+5 0.9477
HOMO-1 -> LUMO+1 -0.2426
rsp_drv.plot(rsp_results)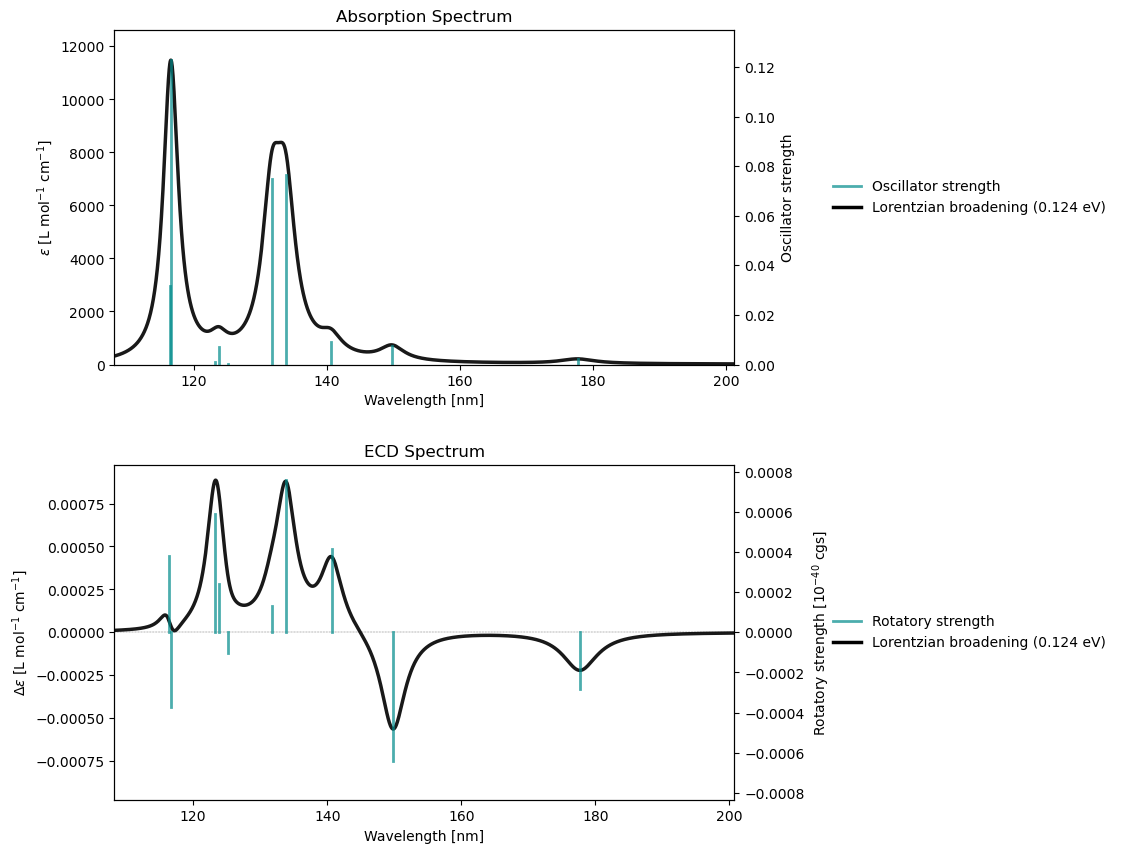
From an h5 file
rsp_results = vlx.read_results("../output_files/alanine-ecd.h5", label="rsp")
rsp_drv.plot_ecd(rsp_results)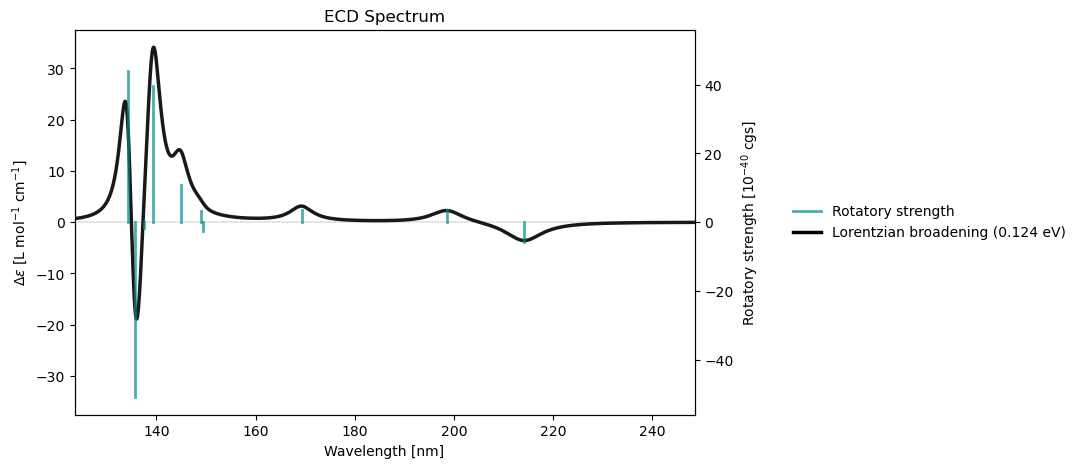
Normal modes¶
From a notebook cell
molecule = vlx.Molecule.read_smiles('CCO')
basis = vlx.MolecularBasis.read(molecule, 'def2-svp')
scf_drv = vlx.ScfRestrictedDriver()
scf_drv.xcfun = 'b3lyp'
scf_results = scf_drv.compute(molecule, basis)
vib_drv = vlx.VibrationalAnalysis(scf_drv)
vib_results = vib_drv.compute(molecule, basis)
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Kohn-Sham
Initial Guess Model : Superposition of Atomic Densities
Convergence Accelerator : Two Level Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Nuclear repulsion energy: 81.6235499154 a.u.
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Molecular grid with 115688 points generated in 0.04 sec.
* Info * Overlap matrix computed in 0.01 sec.
* Info * Kinetic energy matrix computed in 0.00 sec.
* Info * Nuclear potential matrix computed in 0.00 sec.
* Info * Orthogonalization matrix computed in 0.00 sec.
* Info * Starting Reduced Basis SCF calculation...
* Info * ...done. SCF energy in reduced basis set: -153.879535899792 a.u. Time: 0.09 sec.
* Info * Overlap matrix computed in 0.00 sec.
* Info * Kinetic energy matrix computed in 0.01 sec.
* Info * Nuclear potential matrix computed in 0.00 sec.
* Info * Orthogonalization matrix computed in 0.00 sec.
Iter. | Kohn-Sham Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -154.914794427481 0.0000000000 0.25018176 0.01667976 0.00000000
2 -154.917119939846 -0.0023255124 0.19462327 0.01472408 0.14964109
3 -154.921762513318 -0.0046425735 0.03453569 0.00229111 0.06939529
4 -154.921897572842 -0.0001350595 0.00585472 0.00048442 0.01097434
5 -154.921899814084 -0.0000022412 0.00321337 0.00025903 0.00264134
6 -154.921900756934 -0.0000009429 0.00024575 0.00001516 0.00096060
7 -154.921900763193 -0.0000000063 0.00007656 0.00000428 0.00009534
8 -154.921900763889 -0.0000000007 0.00001457 0.00000103 0.00002510
9 -154.921900763915 -0.0000000000 0.00000174 0.00000014 0.00000525
10 -154.921900763915 -0.0000000000 0.00000022 0.00000002 0.00000085
*** SCF converged in 10 iterations. Time: 1.18 sec.
Spin-Restricted Kohn-Sham:
--------------------------
Total Energy : -154.9219007639 a.u.
Electronic Energy : -236.5454506793 a.u.
Nuclear Repulsion Energy : 81.6235499154 a.u.
------------------------------------
Gradient Norm : 0.0000002248 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
Spin Restricted Orbitals
------------------------
Molecular Orbital No. 9:
--------------------------
Occupation: 2.000 Energy: -0.40059 a.u.
( 1 C 1p+1: -0.30) ( 2 C 1p+1: 0.28) ( 2 C 1p0 : -0.16)
( 2 C 2p+1: 0.16) ( 3 O 3s : 0.18) ( 3 O 1p+1: 0.15)
( 3 O 1p0 : 0.33) ( 3 O 2p0 : 0.20)
Molecular Orbital No. 10:
--------------------------
Occupation: 2.000 Energy: -0.37987 a.u.
( 1 C 1p0 : 0.40) ( 6 H 1s : -0.38) ( 6 H 2s : -0.19)
Molecular Orbital No. 11:
--------------------------
Occupation: 2.000 Energy: -0.36361 a.u.
( 1 C 1p-1: -0.34) ( 1 C 2p-1: -0.15) ( 2 C 1p-1: 0.17)
( 3 O 1p-1: 0.20) ( 4 H 1s : -0.28) ( 5 H 1s : 0.28)
Molecular Orbital No. 12:
--------------------------
Occupation: 2.000 Energy: -0.32392 a.u.
( 1 C 3s : -0.16) ( 1 C 1p+1: 0.26) ( 2 C 1p+1: -0.26)
( 2 C 1p0 : -0.17) ( 3 O 3s : 0.24) ( 3 O 1p+1: 0.30)
( 3 O 1p0 : 0.28) ( 3 O 2p+1: 0.19) ( 3 O 2p0 : 0.20)
( 7 H 1s : 0.16) ( 8 H 1s : 0.16) ( 9 H 1s : -0.20)
Molecular Orbital No. 13:
--------------------------
Occupation: 2.000 Energy: -0.26363 a.u.
( 2 C 1p-1: 0.18) ( 3 O 1p+1: 0.19) ( 3 O 1p-1: -0.52)
( 3 O 2p-1: -0.39) ( 7 H 1s : -0.23) ( 7 H 2s : -0.17)
( 8 H 1s : 0.23) ( 8 H 2s : 0.17)
Molecular Orbital No. 14:
--------------------------
Occupation: 0.000 Energy: 0.04506 a.u.
( 1 C 3s : -0.50) ( 1 C 2p+1: -0.23) ( 2 C 3s : -1.29)
( 2 C 1p0 : 0.17) ( 2 C 2p+1: 0.20) ( 2 C 2p0 : 0.32)
( 3 O 3s : -0.55) ( 3 O 1p+1: 0.18) ( 3 O 2p+1: 0.27)
( 4 H 2s : 0.22) ( 5 H 2s : 0.22) ( 6 H 2s : 0.51)
( 7 H 2s : 0.73) ( 8 H 2s : 0.73) ( 9 H 2s : 1.00)
Molecular Orbital No. 15:
--------------------------
Occupation: 0.000 Energy: 0.08189 a.u.
( 1 C 2s : -0.16) ( 1 C 3s : -2.26) ( 1 C 2p+1: -0.26)
( 2 C 3s : -0.31) ( 2 C 2p+1: 0.39) ( 3 O 3s : 0.44)
( 3 O 1p+1: -0.17) ( 3 O 2p+1: -0.26) ( 4 H 2s : 0.98)
( 5 H 2s : 0.98) ( 6 H 2s : 0.92) ( 7 H 2s : 0.44)
( 8 H 2s : 0.44) ( 9 H 2s : -0.68)
Molecular Orbital No. 16:
--------------------------
Occupation: 0.000 Energy: 0.11463 a.u.
( 1 C 1p-1: 0.21) ( 1 C 2p+1: -0.19) ( 1 C 2p-1: 0.53)
( 1 C 2p0 : 0.15) ( 2 C 1p-1: 0.30) ( 2 C 2p+1: -0.29)
( 2 C 2p-1: 0.80) ( 2 C 2p0 : 0.23) ( 4 H 2s : -0.92)
( 5 H 2s : 0.92) ( 7 H 2s : 1.25) ( 8 H 2s : -1.25)
Molecular Orbital No. 17:
--------------------------
Occupation: 0.000 Energy: 0.12054 a.u.
( 1 C 3s : 2.12) ( 1 C 2p0 : 0.18) ( 2 C 2s : -0.18)
( 2 C 3s : -2.39) ( 2 C 2p0 : 0.37) ( 3 O 3s : 0.45)
( 3 O 2p+1: -0.16) ( 4 H 2s : -0.89) ( 5 H 2s : -0.89)
( 6 H 2s : -0.39) ( 7 H 2s : 1.27) ( 8 H 2s : 1.27)
( 9 H 2s : -0.58)
Molecular Orbital No. 18:
--------------------------
Occupation: 0.000 Energy: 0.13626 a.u.
( 1 C 3s : 0.33) ( 1 C 1p0 : -0.36) ( 1 C 2p+1: -0.25)
( 1 C 2p-1: 0.27) ( 1 C 2p0 : -1.24) ( 2 C 3s : -0.54)
( 2 C 2p+1: 0.16) ( 2 C 2p0 : 0.34) ( 4 H 2s : 0.77)
( 5 H 2s : 0.77) ( 6 H 2s : -1.98) ( 7 H 2s : 0.38)
( 8 H 2s : 0.38)
Ground State Dipole Moment
----------------------------
X : -0.459683 a.u. -1.168397 Debye
Y : -0.054679 a.u. -0.138980 Debye
Z : -0.392837 a.u. -0.998493 Debye
Total : 0.607140 a.u. 1.543197 Debye
Vibrational Analysis Driver
=============================
The following will be computed:
- Vibrational frequencies and normal modes
- Force constants
- IR intensities
SCF Hessian Driver
====================
Hessian Type : Analytical
* Info * Computing analytical Hessian...
Reference: P. Deglmann, F. Furche, R. Ahlrichs, Chem. Phys. Lett. 2002, 362, 511-518.
Coupled-Perturbed Kohn-Sham Solver Setup
==========================================
Solver Type : Iterative Subspace Algorithm
Max. Number of Iterations : 150
Convergence Threshold : 1.0e-04
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Molecular grid with 115688 points generated in 0.04 sec.
* Info * CPHF/CPKS integral derivatives computed in 2.14 sec.
* Info * CPHF/CPKS right-hand side computed in 2.54 sec.
* Info * Processing 24 Fock builds...
* Info * 24 trial vectors in reduced space
* Info * 461.85 kB of memory used for subspace procedure on the master node
* Info * 4.64 GB of memory available for the solver on the master node
*** Iteration: 1 * Residuals (Max,Min): 4.24e-01 and 1.13e-01
* Info * Processing 24 Fock builds...
* Info * 48 trial vectors in reduced space
* Info * 756.38 kB of memory used for subspace procedure on the master node
* Info * 4.65 GB of memory available for the solver on the master node
*** Iteration: 2 * Residuals (Max,Min): 3.72e-02 and 1.02e-02
* Info * Processing 24 Fock builds...
* Info * 72 trial vectors in reduced space
* Info * 1.05 MB of memory used for subspace procedure on the master node
* Info * 4.63 GB of memory available for the solver on the master node
*** Iteration: 3 * Residuals (Max,Min): 3.53e-03 and 6.61e-04
* Info * Processing 17 Fock builds...
* Info * 89 trial vectors in reduced space
* Info * 1.30 MB of memory used for subspace procedure on the master node
* Info * 4.57 GB of memory available for the solver on the master node
*** Iteration: 4 * Residuals (Max,Min): 2.12e-04 and 4.42e-05
* Info * Processing 18 Fock builds...
* Info * 107 trial vectors in reduced space
* Info * 1.65 MB of memory used for subspace procedure on the master node
* Info * 4.66 GB of memory available for the solver on the master node
*** Iteration: 5 * Residuals (Max,Min): 9.49e-05 and 5.23e-06
*** Coupled-Perturbed Kohn-Sham converged in 5 iterations. Time: 13.58 sec
* Info * First order derivative contributions to the Hessian computed in 0.00 sec.
* Info * Second order derivative contributions to the Hessian computed in 11.60 sec.
*** Time spent in Hessian calculation: 25.21 sec ***
Free Energy Analysis
======================
Note: Rotational symmetry is set to 1 regardless of true symmetry
No Imaginary Frequencies
Free energy contributions calculated at @ 298.15 K:
Zero-point vibrational energy: 50.1783 kcal/mol
H (Trans + Rot + Vib = Tot): 1.4812 + 0.8887 + 0.6068 = 2.9767 kcal/mol
S (Trans + Rot + Vib = Tot): 37.4339 + 22.3009 + 2.8368 = 62.5716 cal/mol/K
TS (Trans + Rot + Vib = Tot): 11.1609 + 6.6490 + 0.8458 = 18.6557 kcal/mol
Ground State Electronic Energy : E0 = -154.92190076 au ( -97214.9605 kcal/mol)
Free Energy Correction (Harmonic) : ZPVE + [H-TS]_T,R,V = 0.05497819 au ( 34.4993 kcal/mol)
Gibbs Free Energy (Harmonic) : E0 + ZPVE + [H-TS]_T,R,V = -154.86692257 au ( -97180.4611 kcal/mol)
Vibrational Analysis
======================
* Info * The 5 dominant normal modes are printed below.
Vibrational Mode 3
----------------------------------------------------
Harmonic frequency: 671.45 cm**-1
Reduced mass: 1.0931 amu
Force constant: 0.2904 mdyne/A
IR intensity: 114.9188 km/mol
Normal mode:
X Y Z
1 C 0.0098 -0.0267 -0.0077
2 C -0.0141 0.0387 0.0112
3 O -0.0202 0.0552 0.0159
4 H 0.0988 -0.0468 -0.0023
5 H -0.0485 -0.0906 -0.0374
6 H 0.0063 -0.0171 -0.0049
7 H 0.0160 0.0085 0.0465
8 H -0.0257 0.0179 -0.0389
9 H 0.3254 -0.8902 -0.2569
Vibrational Mode 7
----------------------------------------------------
Harmonic frequency: 1181.12 cm**-1
Reduced mass: 2.9121 amu
Force constant: 2.3936 mdyne/A
IR intensity: 44.6017 km/mol
Normal mode:
X Y Z
1 C -0.0253 -0.0325 0.0806
2 C 0.0274 0.0956 -0.2967
3 O -0.0127 -0.0656 0.2114
4 H 0.0229 0.1410 -0.1576
5 H 0.0753 -0.0023 -0.1989
6 H -0.3575 -0.1280 -0.0093
7 H 0.2272 0.1724 -0.3924
8 H 0.2129 0.2117 -0.3810
9 H -0.0047 -0.1051 0.3583
Vibrational Mode 10
----------------------------------------------------
Harmonic frequency: 1344.62 cm**-1
Reduced mass: 1.1757 amu
Force constant: 1.2524 mdyne/A
IR intensity: 111.5237 km/mol
Normal mode:
X Y Z
1 C -0.0188 0.0105 -0.0601
2 C -0.0107 -0.0264 0.0779
3 O -0.0301 0.0026 -0.0473
4 H -0.0210 -0.1270 0.1160
5 H -0.0726 0.0141 0.1567
6 H 0.1262 0.0524 -0.0218
7 H 0.3657 0.1524 -0.2090
8 H 0.3408 0.2208 -0.1893
9 H 0.0904 -0.1648 0.6856
Vibrational Mode 16
----------------------------------------------------
Harmonic frequency: 2921.01 cm**-1
Reduced mass: 1.0548 amu
Force constant: 5.3027 mdyne/A
IR intensity: 77.3339 km/mol
Normal mode:
X Y Z
1 C -0.0001 -0.0017 0.0057
2 C -0.0474 -0.0046 -0.0441
3 O 0.0012 0.0013 -0.0031
4 H 0.0018 0.0033 0.0051
5 H 0.0043 -0.0035 0.0032
6 H 0.0033 0.0214 -0.0700
7 H 0.0768 0.5440 0.4397
8 H 0.4629 -0.5125 0.1349
9 H -0.0026 0.0012 -0.0075
Vibrational Mode 18
----------------------------------------------------
Harmonic frequency: 2963.37 cm**-1
Reduced mass: 1.1092 amu
Force constant: 5.7387 mdyne/A
IR intensity: 71.5678 km/mol
Normal mode:
X Y Z
1 C 0.0049 -0.0134 -0.0039
2 C -0.0314 0.0859 0.0248
3 O 0.0000 -0.0000 -0.0000
4 H 0.0137 0.0865 0.0729
5 H -0.0753 0.0821 -0.0243
6 H 0.0015 -0.0043 -0.0012
7 H -0.0968 -0.5319 -0.4360
8 H 0.4706 -0.4910 0.1410
9 H 0.0017 -0.0047 -0.0013
vib_drv.plot(vib_results)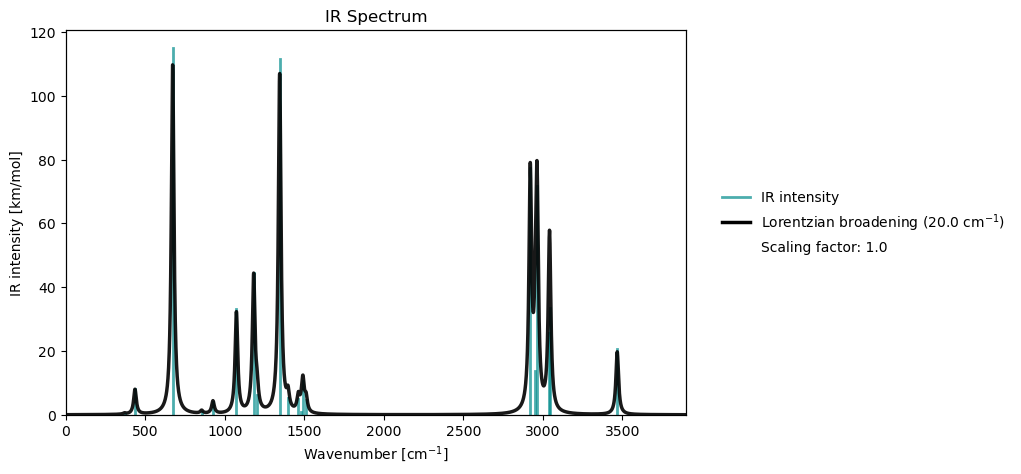
vib_drv.animate(vib_results, mode=14)Loading...
From an h5 file
vib_results = vlx.read_results("../output_files/acro-raman.h5", label="vib")
vib_drv.plot_raman(vib_results)
vib_drv.animate(vib_results, mode=14)Loading...