Conformational search aims to identify the possible three-dimensional arrangements of a molecule by exploring its torsional degrees of freedom. For small and rigid molecules, a systematic enumeration of all rotatable bond combinations is feasible and guarantees complete coverage of the conformational space. However, as the number of rotatable bonds grows, the combinatorial explosion of possible conformations makes systematic search computationally intractable for larger and more flexible systems.
For small molecules, VeloxChem provides the ConformerGenerator class, which performs a systematic search by enumerating all combinations of rotatable bond angles, followed by MM energy minimization of each generated structure, yielding a ranked list of low-energy conformers.
For larger and more flexible systems, VeloxChem offers an MD-based conformational sampling approach through the conformational_sampling method of the OpenMMDynamics class. By running a high-temperature MD simulation, the molecule can overcome torsional barriers and broadly explore its conformational space. Snapshots collected along the trajectory are energy-minimized and, optionally, filtered to retain only unique conformers, providing a diverse and representative ensemble of structures.
Systematic Search¶
import veloxchem as vlx
molecule = vlx.Molecule.read_smiles(
"CC1([C@@H](N2[C@H](S1)[C@@H](C2=O)NC(=O)CC3=CC=CC=C3)C(=O)O)C")
molecule.show(atom_indices=True)The ConformerGenerator class can generate all possible conformations and minimize them with MM.
conf = vlx.ConformerGenerator()
conformers_dict = conf.generate(molecule)
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Hartree-Fock
Initial Guess Model : Superposition of Atomic Densities
Convergence Accelerator : Two Level Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
* Info * Starting Reduced Basis SCF calculation...
* Info * ...done. SCF energy in reduced basis set: -1421.698355462260 a.u. Time: 5.06 sec.
Iter. | Hartree-Fock Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -1422.145650692370 0.0000000000 0.34583788 0.01240430 0.00000000
2 -1422.159172976852 -0.0135222845 0.14071348 0.00699143 0.20177360
3 -1422.161099225940 -0.0019262491 0.04775721 0.00193235 0.06270231
4 -1422.161350895974 -0.0002516700 0.01296397 0.00052355 0.01722927
5 -1422.161374814897 -0.0000239189 0.00355159 0.00015290 0.00718887
6 -1422.161377216099 -0.0000024012 0.00127657 0.00005056 0.00251591
7 -1422.161377569155 -0.0000003531 0.00048027 0.00002830 0.00086756
8 -1422.161377651816 -0.0000000827 0.00022962 0.00001169 0.00046344
9 -1422.161377671894 -0.0000000201 0.00011290 0.00000638 0.00018043
10 -1422.161377679166 -0.0000000073 0.00004414 0.00000272 0.00012718
11 -1422.161377680369 -0.0000000012 0.00001746 0.00000116 0.00005339
12 -1422.161377680515 -0.0000000001 0.00000662 0.00000048 0.00001888
13 -1422.161377680539 -0.0000000000 0.00000292 0.00000016 0.00000698
14 -1422.161377680542 -0.0000000000 0.00000135 0.00000009 0.00000265
15 -1422.161377680543 -0.0000000000 0.00000055 0.00000002 0.00000128
*** SCF converged in 15 iterations. Time: 31.43 sec.
Spin-Restricted Hartree-Fock:
-----------------------------
Total Energy : -1422.1613776805 a.u.
Electronic Energy : -3526.5881020333 a.u.
Nuclear Repulsion Energy : 2104.4267243528 a.u.
------------------------------------
Gradient Norm : 0.0000005546 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
RESP Charges Driver Setup
===========================
Number of Conformers : 1
Number of Layers : 4
Points per Square Angstrom : 1.0
Total Number of Grid Points : 1669
First Stage Fit
-----------------
Restraint Strength : 0.0005
Restrained Hydrogens : No
Max. Number of Iterations : 50
Convergence Threshold (a.u.) : 1e-06
*** Charge fitting converged in 16 iterations.
No. | Atom | Constraints | Charges (a.u.)
--------------------------------------------
1 C -0.422043
2 C 0.203275
3 C 0.002507
4 N -0.315237
5 C 0.150969
6 S -0.215332
7 C 0.043108
8 C 0.454156
9 O -0.477416
10 N -0.507137
11 C 0.507132
12 O -0.511646
13 C -0.084649
14 C 0.040186
15 C -0.199950
16 C -0.164121
17 C -0.107273
18 C -0.177760
19 C -0.085509
20 C 0.695366
21 O -0.563140
22 O -0.707575
23 C -0.304244
24 H 0.133756
25 H 0.110597
26 H 0.136174
27 H 0.160018
28 H 0.092295
29 H 0.130960
30 H 0.314466
31 H 0.091331
32 H 0.039812
33 H 0.154838
34 H 0.151406
35 H 0.135216
36 H 0.151046
37 H 0.118912
38 H 0.512588
39 H 0.133628
40 H 0.076940
41 H 0.102350
--------------------------------------------
Total Charge : 0.000000
Fit Quality
-------------
Relative Root-Mean-Square Error : 0.110055
Second Stage Fit
------------------
Restraint Strength : 0.001
Restrained Hydrogens : No
Max. Number of Iterations : 50
Convergence Threshold (a.u.) : 1e-06
*** Charge fitting converged in 16 iterations.
No. | Atom | Frozen | Constraints | Charges (a.u.)
----------------------------------------------------
1 C No -0.382528
2 C Yes 0.203275
3 C Yes 0.002507
4 N Yes -0.315237
5 C Yes 0.150969
6 S Yes -0.215332
7 C Yes 0.043108
8 C Yes 0.454156
9 O Yes -0.477416
10 N Yes -0.507137
11 C Yes 0.507132
12 O Yes -0.511646
13 C No -0.088868
14 C Yes 0.040186
15 C Yes -0.199950
16 C Yes -0.164121
17 C Yes -0.107273
18 C Yes -0.177760
19 C Yes -0.085509
20 C Yes 0.695366
21 O Yes -0.563140
22 O Yes -0.707575
23 C No -0.395778
24 H No 0.116020
25 H No 24 0.116020
26 H No 25 0.116020
27 H Yes 0.160018
28 H Yes 0.092295
29 H Yes 0.130960
30 H Yes 0.314466
31 H No 0.072382
32 H No 31 0.072382
33 H Yes 0.154838
34 H Yes 0.151406
35 H Yes 0.135216
36 H Yes 0.151046
37 H Yes 0.118912
38 H Yes 0.512588
39 H No 0.129334
40 H No 39 0.129334
41 H No 40 0.129334
----------------------------------------------------
Total Charge : 0.000000
Fit Quality
-------------
Relative Root-Mean-Square Error : 0.126732
Reference:
J. Phys. Chem. 1993, 97, 10269-10280.
* Info * 36 conformers will be generated.
* Info * 36 conformers generated in 0.32 sec
* Info * Energy minimization of 36 conformers took 1.23 sec
* Info * Global minimum energy: -247.407 kJ/mol
* Info * 11 conformers remain after removal of duplicate conformers.
* Info * Total time spent in generating conformers: 39.23 sec
The lowest conformer can be displayed
conf.show_global_minimum()Global minimum conformer with energy -247.407 kJ/mol
And more conformers can be diplayed
conf.show_conformers(number=3)Conformer 1 with energy -247.407 kJ/mol
Conformer 2 with energy -243.576 kJ/mol
Conformer 3 with energy -242.980 kJ/mol
Extract conformers from an MD simulation¶
molecule = vlx.Molecule.read_xyz_file("../input_files/tq-polymer.xyz")
molecule.show()partial_charges = [
-0.232669, -0.048234, -0.007686, 0.100183, 0.386987,
-0.249451, 0.194951, 0.114678, 0.062361, 0.005422,
-0.107660, -0.198262, 0.016200, -0.090324, -0.235291,
0.062137, 0.419456, -0.531796, 0.139008, 0.164115,
-0.086069, -0.044513, 0.199556, -0.170296, -0.180157,
-0.113322, 0.158106, -0.081966, -0.128774, -0.239557,
-0.136771, 0.301582, -0.203020, 0.138342, 0.136587,
-0.316948, -0.031826, 0.137154, 0.118999, 0.161926,
0.130632, 0.173826, -0.332761, -0.005770, 0.079527,
0.079527, 0.065962, 0.065962, -0.242085, -0.087463,
0.012935, 0.061502, 0.266088, -0.001114, 0.103725,
0.180948, 0.015843, -0.123259, -0.242504, 0.056159,
-0.118009, -0.247948, 0.196117, 0.205390, -0.298929,
0.084877, 0.079527, 0.158266, -0.146844, -0.040754,
0.251020, -0.203210, -0.160584, -0.153068, 0.159255,
-0.052933, -0.119071, -0.248423, -0.079409, 0.161470,
-0.134947, 0.149941, 0.170980, -0.324275, -0.042230,
0.077237, 0.144781, 0.111871, 0.146494, 0.118468,
-0.259568, -0.020448, 0.079707, 0.079707, 0.065165,
0.065165, 0.065962, 0.065165, 0.079707, 0.091273,
-0.257720, -0.061333, 0.006290, 0.053719, 0.241539,
0.008275, 0.177544, 0.136687, 0.102227, 0.036805,
-0.167956, -0.170007, 0.026697, -0.121728, -0.219001,
0.179470, 0.218907, -0.269483, 0.130723, 0.169499,
-0.141246, -0.045503, 0.231592, -0.203064, -0.149092,
-0.160352, 0.156878, -0.087277, -0.117300, -0.253518,
-0.105665, 0.201677, -0.129198, 0.154380, 0.144414,
-0.311560, -0.050335, 0.169505, 0.117806, 0.078577,
0.104835, 0.177924, -0.277724, -0.030888, 0.083872,
0.083872, 0.071197, 0.071197, -0.205491, -0.105325,
0.004215, 0.071387, 0.205212, 0.002548, 0.100560,
0.158667, 0.021198, -0.109115, -0.258546, 0.071271,
-0.137911, -0.231297, 0.150851, 0.232605, -0.255901,
0.069410, 0.083872, 0.165216, -0.118096, -0.035060,
0.232363, -0.199482, -0.160185, -0.163606, 0.159908,
-0.075661, -0.119060, -0.255872, -0.099171, 0.211171,
-0.146255, 0.167897, 0.176623, -0.312825, -0.059878,
0.072443, 0.144852, 0.119482, 0.152389, 0.115928,
-0.288919, -0.023122, 0.084877, 0.084877, 0.069835,
0.069835, 0.071197, 0.069835, -0.277164, -0.038921,
0.002052, 0.076138, 0.220403, 0.008773, 0.186555,
0.122042, 0.064124, 0.006807, -0.102455, -0.211713,
0.025525, -0.100865, -0.225711, 0.134458, 0.220855,
-0.239842, 0.146481, 0.157469, -0.084636, -0.038788,
0.191114, -0.167495, -0.173886, -0.139361, 0.158124,
-0.115619, -0.110832, -0.246506, -0.111697, 0.200255,
-0.102728, 0.147022, 0.137019, -0.311401, -0.028076,
0.157916, 0.121854, 0.070941, 0.097209, 0.174835,
-0.277731, -0.058498, 0.078470, 0.078470, 0.079650,
0.079650, -0.239752, -0.093417, 0.013395, 0.065722,
0.252234, 0.002507, 0.097142, 0.176412, 0.012295,
-0.109919, -0.253222, 0.062514, -0.121338, -0.239217,
0.179990, 0.225267, -0.293594, 0.084407, 0.078470,
0.163374, -0.156555, -0.027891, 0.267086, -0.220163,
-0.153943, -0.138332, 0.156524, -0.068274, -0.107757,
-0.248650, -0.083734, 0.150618, -0.128876, 0.155916,
0.171802, -0.332583, -0.033320, 0.071492, 0.147678,
0.114573, 0.140265, 0.115307, -0.249021, -0.028279,
0.077074, 0.077074, 0.068191, 0.068191, 0.079650,
0.068191, 0.077074, 0.085617, -0.262181, -0.058425,
0.004567, 0.054389, 0.245865, 0.006966, 0.178357,
0.134007, 0.098000, 0.031254, -0.152193, -0.172248,
0.024457, -0.114391, -0.218354, 0.183237, 0.211562,
-0.273990, 0.125284, 0.158656, -0.144065, -0.033540,
0.216218, -0.188581, -0.155284, -0.157807, 0.156199,
-0.078857, -0.117411, -0.254610, -0.097991, 0.198924,
-0.133192, 0.153092, 0.142005, -0.306650, -0.071477,
0.168597, 0.113979, 0.075060, 0.107961, 0.177382,
-0.281830, -0.022927, 0.092650, 0.092650, 0.069410,
0.069410, -0.234824, -0.088279, -0.010610, 0.098104,
0.235745, -0.003237, 0.091918, 0.181039, -0.033203,
-0.007199, -0.314039, -0.121585, -0.064129, -0.269034,
0.181444, 0.222313, -0.282548, 0.200030, 0.092650,
0.175556, -0.124800, -0.033037, 0.242958, -0.205747,
-0.157096, -0.151879, 0.158719, -0.061573, -0.112321,
-0.250936, -0.103306, 0.194760, -0.137261, 0.151524,
0.175522, -0.320888, -0.048925, 0.069816, 0.146477,
0.121450, 0.141797, 0.115735, -0.282016, -0.026049,
0.084407, 0.084407, 0.070941, 0.070941]
ff_gen = vlx.MMForceFieldGenerator()
ff_gen.partial_charges = partial_charges
ff_gen.create_topology(molecule)
opm_dyn = vlx.OpenMMDynamics()
opm_dyn.create_system_from_molecule(molecule,
ff_gen,
filename='tq-polymer',
residue_name='MOL')* Info * Sum of partial charges is not a whole number.
* Info * Compensating by removing 4.000e-06 from the largest charge.
* Info * Using GAFF (v2.11) parameters.
Reference: J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman, D. A. Case, J. Comput. Chem. 2004,
25, 1157-1174.
* Info * Updated bond length 10-14 (cc-ss) to 0.169 nm
* Info * Updated bond length 13-14 (cc-ss) to 0.168 nm
* Info * Updated bond length 36-37 (os-c3) to 0.138 nm
* Info * Updated bond length 43-44 (os-c3) to 0.138 nm
* Info * Updated bond length 51-57 (ca-cc) to 0.151 nm
* Info * Updated bond length 57-61 (cc-ss) to 0.169 nm
* Info * Updated bond length 60-61 (cd-ss) to 0.168 nm
* Info * Updated bond length 60-106 (cd-ca) to 0.151 nm
* Info * Updated bond length 84-85 (os-c3) to 0.138 nm
* Info * Updated bond length 91-92 (os-c3) to 0.138 nm
* Info * Updated bond length 103-110 (ca-cc) to 0.151 nm
* Info * Updated bond length 110-114 (cc-ss) to 0.169 nm
* Info * Updated bond length 113-114 (cc-ss) to 0.169 nm
* Info * Updated bond length 113-154 (cc-ca) to 0.151 nm
* Info * Updated bond length 136-137 (os-c3) to 0.138 nm
* Info * Updated bond length 143-144 (os-c3) to 0.138 nm
* Info * Updated bond length 151-157 (ca-cc) to 0.151 nm
* Info * Updated bond length 157-161 (cc-ss) to 0.169 nm
* Info * Updated bond length 160-161 (cc-ss) to 0.169 nm
* Info * Updated bond length 160-204 (cc-ca) to 0.151 nm
* Info * Updated bond length 184-185 (os-c3) to 0.138 nm
* Info * Updated bond length 191-192 (os-c3) to 0.138 nm
* Info * Updated bond length 201-208 (ca-cc) to 0.151 nm
* Info * Updated bond length 208-212 (cc-ss) to 0.169 nm
* Info * Updated bond length 211-212 (cc-ss) to 0.169 nm
* Info * Updated bond length 211-252 (cc-ca) to 0.151 nm
* Info * Updated bond length 234-235 (os-c3) to 0.138 nm
* Info * Updated bond length 241-242 (os-c3) to 0.138 nm
* Info * Updated bond length 249-255 (ca-cc) to 0.151 nm
* Info * Updated bond length 255-259 (cc-ss) to 0.169 nm
* Info * Updated bond length 258-259 (cc-ss) to 0.169 nm
* Info * Updated bond length 258-304 (cc-ca) to 0.151 nm
* Info * Updated bond length 282-283 (os-c3) to 0.138 nm
* Info * Updated bond length 289-290 (os-c3) to 0.138 nm
* Info * Updated bond length 301-308 (ca-cc) to 0.151 nm
* Info * Updated bond length 308-312 (cc-ss) to 0.169 nm
* Info * Updated bond length 311-312 (cc-ss) to 0.168 nm
* Info * Updated bond length 334-335 (os-c3) to 0.138 nm
* Info * Updated bond length 341-342 (os-c3) to 0.138 nm
* Info * Updated bond length 355-359 (cc-ss) to 0.169 nm
* Info * Updated bond length 358-359 (cd-ss) to 0.168 nm
* Info * Updated bond length 382-383 (os-c3) to 0.138 nm
* Info * Updated bond length 389-390 (os-c3) to 0.138 nm
* Info * Updated bond angle 3-10-11 (ca-cc-cd) to 129.147 deg
* Info * Updated bond angle 12-13-54 (cd-cc-ca) to 126.821 deg
* Info * Updated bond angle 51-57-58 (ca-cc-cd) to 129.494 deg
* Info * Updated bond angle 59-60-61 (cd-cd-ss) to 105.976 deg
* Info * Updated bond angle 59-60-106 (cd-cd-ca) to 127.140 deg
* Info * Updated bond angle 103-110-111 (ca-cc-cd) to 129.577 deg
* Info * Updated bond angle 112-113-154 (cd-cc-ca) to 127.147 deg
* Info * Updated bond angle 151-157-158 (ca-cc-cd) to 129.113 deg
* Info * Updated bond angle 159-160-204 (cd-cc-ca) to 126.734 deg
* Info * Updated bond angle 201-208-209 (ca-cc-cd) to 128.625 deg
* Info * Updated bond angle 210-211-252 (cd-cc-ca) to 126.063 deg
* Info * Updated bond angle 249-255-256 (ca-cc-cd) to 128.947 deg
* Info * Updated bond angle 257-258-304 (cd-cc-ca) to 126.537 deg
* Info * Updated bond angle 301-308-309 (ca-cc-cd) to 129.285 deg
* Info * Updated bond angle 310-311-352 (cd-cc-ca) to 126.759 deg
* Info * Updated bond angle 349-355-356 (ca-cc-cc) to 128.976 deg
* Info * Updated bond angle 356-355-359 (cc-cc-ss) to 106.576 deg
***********
* Warning * MMForceFieldGenerator: dihedral ca-nb-cq-cp is not available.
***********
***********
* Warning * MMForceFieldGenerator: dihedral ca-nb-cq-cq is not available.
***********
***********
* Warning * MMForceFieldGenerator: dihedral ca-nb-cq-cp is not available.
***********
***********
* Warning * MMForceFieldGenerator: dihedral ca-nb-cq-cq is not available.
***********
***********
* Warning * MMForceFieldGenerator: dihedral ca-nb-cq-cp is not available.
***********
***********
* Warning * MMForceFieldGenerator: dihedral ca-nb-cq-cq is not available.
***********
***********
* Warning * MMForceFieldGenerator: dihedral ca-nb-cq-cp is not available.
***********
***********
* Warning * MMForceFieldGenerator: dihedral ca-nb-cq-cq is not available.
***********
***********
* Warning * MMForceFieldGenerator: dihedral ca-nb-cq-cp is not available.
***********
***********
* Warning * MMForceFieldGenerator: dihedral ca-nb-cq-cq is not available.
***********
***********
* Warning * MMForceFieldGenerator: dihedral ca-nb-cq-cp is not available.
***********
***********
* Warning * MMForceFieldGenerator: dihedral ca-nb-cq-cq is not available.
***********
***********
* Warning * MMForceFieldGenerator: dihedral ca-nb-cq-cp is not available.
***********
***********
* Warning * MMForceFieldGenerator: dihedral ca-nb-cq-cq is not available.
***********
***********
* Warning * MMForceFieldGenerator: dihedral ca-nb-cq-cp is not available.
***********
***********
* Warning * MMForceFieldGenerator: dihedral ca-nb-cq-cq is not available.
***********
* Info * System parameters written to tq-polymer_system.xml
* Info * System coordinates written to tq-polymer_system.pdb
Once a force field has been derived with the MMForceFieldGenerator and the system has been initialized with the OpenMMDynamics class; the conformational_sampling function can be used to extract conformers from the MD simulation at high temperature. The user can define a number of snapshots that will be optimized and save. In addition, those snapshots can be filtered to avoid that the same conformer is present multiple timess by using the option unique_conformers.
conformers_dict = opm_dyn.conformational_sampling(
ensemble='NVT',
temperature=1000,
timestep=2.0,
nsteps=100000,
snapshots=500,
unique_conformers=True,
qm_driver=None,
basis=None,
constraints=None)* Info * Minimized energy: 1645.3622207045555
* Info * Saved coordinates for step 200
* Info * Minimized energy: 1655.1349096298218
* Info * Saved coordinates for step 400
* Info * Minimized energy: 1663.6939027905464
* Info * Saved coordinates for step 600
* Info * Minimized energy: 1671.5768882632256
* Info * Saved coordinates for step 800
* Info * Minimized energy: 1678.4117020368576
* Info * Saved coordinates for step 1000
* Info * Minimized energy: 1685.2487353086472
* Info * Saved coordinates for step 1200
* Info * Minimized energy: 1692.5561211109161
* Info * Saved coordinates for step 1400
* Info * Minimized energy: 1682.5339225232601
* Info * Saved coordinates for step 1600
* Info * Minimized energy: 1686.3644063472748
* Info * Saved coordinates for step 1800
* Info * Minimized energy: 1724.5460814237595
* Info * Saved coordinates for step 2000
* Info * Minimized energy: 1733.2746371924877
* Info * Saved coordinates for step 2200
* Info * Minimized energy: 1708.6360507905483
* Info * Saved coordinates for step 2400
* Info * Minimized energy: 1675.569159924984
* Info * Saved coordinates for step 2600
* Info * Minimized energy: 1690.6314902082086
* Info * Saved coordinates for step 2800
* Info * Minimized energy: 1693.4284328073263
* Info * Saved coordinates for step 3000
* Info * Minimized energy: 1666.7326683849096
* Info * Saved coordinates for step 3200
* Info * Minimized energy: 1630.0430088303983
* Info * Saved coordinates for step 3400
* Info * Minimized energy: 1648.780307635665
* Info * Saved coordinates for step 3600
* Info * Minimized energy: 1638.171914294362
* Info * Saved coordinates for step 3800
* Info * Minimized energy: 1678.3075994551182
* Info * Saved coordinates for step 4000
* Info * Minimized energy: 1650.4017176926136
* Info * Saved coordinates for step 4200
* Info * Minimized energy: 1651.0782541930676
* Info * Saved coordinates for step 4400
* Info * Minimized energy: 1661.2700364589691
* Info * Saved coordinates for step 4600
* Info * Minimized energy: 1649.768322698772
* Info * Saved coordinates for step 4800
* Info * Minimized energy: 1677.2823501229286
* Info * Saved coordinates for step 5000
* Info * Minimized energy: 1731.6282952427864
* Info * Saved coordinates for step 5200
* Info * Minimized energy: 1737.254495024681
* Info * Saved coordinates for step 5400
* Info * Minimized energy: 1740.6430314779282
* Info * Saved coordinates for step 5600
* Info * Minimized energy: 1706.3045947551727
* Info * Saved coordinates for step 5800
* Info * Minimized energy: 1693.4855611920357
* Info * Saved coordinates for step 6000
* Info * Minimized energy: 1693.1725072264671
* Info * Saved coordinates for step 6200
* Info * Minimized energy: 1716.987262904644
* Info * Saved coordinates for step 6400
* Info * Minimized energy: 1672.8852657079697
* Info * Saved coordinates for step 6600
* Info * Minimized energy: 1692.1092131137848
* Info * Saved coordinates for step 6800
* Info * Minimized energy: 1675.3911390006542
* Info * Saved coordinates for step 7000
* Info * Minimized energy: 1684.8245009481907
* Info * Saved coordinates for step 7200
* Info * Minimized energy: 1709.883114695549
* Info * Saved coordinates for step 7400
* Info * Minimized energy: 1698.7981094121933
* Info * Saved coordinates for step 7600
* Info * Minimized energy: 1705.3959839642048
* Info * Saved coordinates for step 7800
* Info * Minimized energy: 1636.011299431324
* Info * Saved coordinates for step 8000
* Info * Minimized energy: 1681.0824654102325
* Info * Saved coordinates for step 8200
* Info * Minimized energy: 1650.6314700245857
* Info * Saved coordinates for step 8400
* Info * Minimized energy: 1652.8125948309898
* Info * Saved coordinates for step 8600
* Info * Minimized energy: 1629.694775223732
* Info * Saved coordinates for step 8800
* Info * Minimized energy: 1648.906716287136
* Info * Saved coordinates for step 9000
* Info * Minimized energy: 1669.111581981182
* Info * Saved coordinates for step 9200
* Info * Minimized energy: 1663.4109607338905
* Info * Saved coordinates for step 9400
* Info * Minimized energy: 1688.0166954398155
* Info * Saved coordinates for step 9600
* Info * Minimized energy: 1713.9452012181282
* Info * Saved coordinates for step 9800
* Info * Minimized energy: 1699.0577293038368
* Info * Saved coordinates for step 10000
* Info * Minimized energy: 1715.3360708355904
* Info * Saved coordinates for step 10200
* Info * Minimized energy: 1696.0485010147095
* Info * Saved coordinates for step 10400
* Info * Minimized energy: 1709.578537940979
* Info * Saved coordinates for step 10600
* Info * Minimized energy: 1749.1806028187275
* Info * Saved coordinates for step 10800
* Info * Minimized energy: 1748.910348534584
* Info * Saved coordinates for step 11000
* Info * Minimized energy: 1736.6516065597534
* Info * Saved coordinates for step 11200
* Info * Minimized energy: 1694.7871108055115
* Info * Saved coordinates for step 11400
* Info * Minimized energy: 1681.556333631277
* Info * Saved coordinates for step 11600
* Info * Minimized energy: 1687.008661299944
* Info * Saved coordinates for step 11800
* Info * Minimized energy: 1699.6191420555115
* Info * Saved coordinates for step 12000
* Info * Minimized energy: 1690.333439052105
* Info * Saved coordinates for step 12200
* Info * Minimized energy: 1724.9507036209106
* Info * Saved coordinates for step 12400
* Info * Minimized energy: 1661.7784758806229
* Info * Saved coordinates for step 12600
* Info * Minimized energy: 1660.5185687541962
* Info * Saved coordinates for step 12800
* Info * Minimized energy: 1648.755928516388
* Info * Saved coordinates for step 13000
* Info * Minimized energy: 1653.3355915546417
* Info * Saved coordinates for step 13200
* Info * Minimized energy: 1665.3462694883347
* Info * Saved coordinates for step 13400
* Info * Minimized energy: 1665.107570052147
* Info * Saved coordinates for step 13600
* Info * Minimized energy: 1674.5675617456436
* Info * Saved coordinates for step 13800
* Info * Minimized energy: 1681.1188523769379
* Info * Saved coordinates for step 14000
* Info * Minimized energy: 1681.7083317041397
* Info * Saved coordinates for step 14200
* Info * Minimized energy: 1674.167928159237
* Info * Saved coordinates for step 14400
* Info * Minimized energy: 1672.8461926579475
* Info * Saved coordinates for step 14600
* Info * Minimized energy: 1665.5922718644142
* Info * Saved coordinates for step 14800
* Info * Minimized energy: 1688.978281199932
* Info * Saved coordinates for step 15000
* Info * Minimized energy: 1709.3906506299973
* Info * Saved coordinates for step 15200
* Info * Minimized energy: 1689.3471912145615
* Info * Saved coordinates for step 15400
* Info * Minimized energy: 1680.5954148769379
* Info * Saved coordinates for step 15600
* Info * Minimized energy: 1656.9508278369904
* Info * Saved coordinates for step 15800
* Info * Minimized energy: 1686.0509151220322
* Info * Saved coordinates for step 16000
* Info * Minimized energy: 1663.9452222585678
* Info * Saved coordinates for step 16200
* Info * Minimized energy: 1676.4143463373184
* Info * Saved coordinates for step 16400
* Info * Minimized energy: 1666.334671497345
* Info * Saved coordinates for step 16600
* Info * Minimized energy: 1692.4184294939041
* Info * Saved coordinates for step 16800
* Info * Minimized energy: 1695.341358423233
* Info * Saved coordinates for step 17000
* Info * Minimized energy: 1706.4967346191406
* Info * Saved coordinates for step 17200
* Info * Minimized energy: 1709.3664041757584
* Info * Saved coordinates for step 17400
* Info * Minimized energy: 1753.173506617546
* Info * Saved coordinates for step 17600
* Info * Minimized energy: 1707.9523171186447
* Info * Saved coordinates for step 17800
* Info * Minimized energy: 1697.2300863265991
* Info * Saved coordinates for step 18000
* Info * Minimized energy: 1740.2638337612152
* Info * Saved coordinates for step 18200
* Info * Minimized energy: 1694.0563625693321
* Info * Saved coordinates for step 18400
* Info * Minimized energy: 1696.633165538311
* Info * Saved coordinates for step 18600
* Info * Minimized energy: 1703.2474281787872
* Info * Saved coordinates for step 18800
* Info * Minimized energy: 1703.0545321702957
* Info * Saved coordinates for step 19000
* Info * Minimized energy: 1698.8086940050125
* Info * Saved coordinates for step 19200
* Info * Minimized energy: 1695.8540288209915
* Info * Saved coordinates for step 19400
* Info * Minimized energy: 1656.8228783607483
* Info * Saved coordinates for step 19600
* Info * Minimized energy: 1707.0986820459366
* Info * Saved coordinates for step 19800
* Info * Minimized energy: 1659.7023705244064
* Info * Saved coordinates for step 20000
* Info * Minimized energy: 1734.5847049951553
* Info * Saved coordinates for step 20200
* Info * Minimized energy: 1721.9142179489136
* Info * Saved coordinates for step 20400
* Info * Minimized energy: 1711.9319866895676
* Info * Saved coordinates for step 20600
* Info * Minimized energy: 1670.9825739264488
* Info * Saved coordinates for step 20800
* Info * Minimized energy: 1708.0616744160652
* Info * Saved coordinates for step 21000
* Info * Minimized energy: 1726.5061413645744
* Info * Saved coordinates for step 21200
* Info * Minimized energy: 1695.1069351434708
* Info * Saved coordinates for step 21400
* Info * Minimized energy: 1687.611922621727
* Info * Saved coordinates for step 21600
* Info * Minimized energy: 1687.6980637907982
* Info * Saved coordinates for step 21800
* Info * Minimized energy: 1666.506633758545
* Info * Saved coordinates for step 22000
* Info * Minimized energy: 1687.8661628961563
* Info * Saved coordinates for step 22200
* Info * Minimized energy: 1708.6668565571308
* Info * Saved coordinates for step 22400
* Info * Minimized energy: 1691.3155268132687
* Info * Saved coordinates for step 22600
* Info * Minimized energy: 1677.5899156332016
* Info * Saved coordinates for step 22800
* Info * Minimized energy: 1687.2134407758713
* Info * Saved coordinates for step 23000
* Info * Minimized energy: 1672.2432813048363
* Info * Saved coordinates for step 23200
* Info * Minimized energy: 1707.3449007868767
* Info * Saved coordinates for step 23400
* Info * Minimized energy: 1685.626139998436
* Info * Saved coordinates for step 23600
* Info * Minimized energy: 1692.5698764324188
* Info * Saved coordinates for step 23800
* Info * Minimized energy: 1675.1652269363403
* Info * Saved coordinates for step 24000
* Info * Minimized energy: 1694.4102446436882
* Info * Saved coordinates for step 24200
* Info * Minimized energy: 1654.9567162394524
* Info * Saved coordinates for step 24400
* Info * Minimized energy: 1630.247096836567
* Info * Saved coordinates for step 24600
* Info * Minimized energy: 1640.1464646458626
* Info * Saved coordinates for step 24800
* Info * Minimized energy: 1653.0759460926056
* Info * Saved coordinates for step 25000
* Info * Minimized energy: 1697.137151002884
* Info * Saved coordinates for step 25200
* Info * Minimized energy: 1682.205605685711
* Info * Saved coordinates for step 25400
* Info * Minimized energy: 1672.6200456023216
* Info * Saved coordinates for step 25600
* Info * Minimized energy: 1666.3365000486374
* Info * Saved coordinates for step 25800
* Info * Minimized energy: 1673.7660074234009
* Info * Saved coordinates for step 26000
* Info * Minimized energy: 1692.559344947338
* Info * Saved coordinates for step 26200
* Info * Minimized energy: 1644.450732409954
* Info * Saved coordinates for step 26400
* Info * Minimized energy: 1687.175436347723
* Info * Saved coordinates for step 26600
* Info * Minimized energy: 1682.4864364862442
* Info * Saved coordinates for step 26800
* Info * Minimized energy: 1617.2019211053848
* Info * Saved coordinates for step 27000
* Info * Minimized energy: 1657.5416536927223
* Info * Saved coordinates for step 27200
* Info * Minimized energy: 1628.0452192425728
* Info * Saved coordinates for step 27400
* Info * Minimized energy: 1617.2558215856552
* Info * Saved coordinates for step 27600
* Info * Minimized energy: 1627.6276527643204
* Info * Saved coordinates for step 27800
* Info * Minimized energy: 1623.1719456911087
* Info * Saved coordinates for step 28000
* Info * Minimized energy: 1643.221106350422
* Info * Saved coordinates for step 28200
* Info * Minimized energy: 1658.3412574529648
* Info * Saved coordinates for step 28400
* Info * Minimized energy: 1621.585027217865
* Info * Saved coordinates for step 28600
* Info * Minimized energy: 1641.3104746341705
* Info * Saved coordinates for step 28800
* Info * Minimized energy: 1644.600160062313
* Info * Saved coordinates for step 29000
* Info * Minimized energy: 1691.8208721280098
* Info * Saved coordinates for step 29200
* Info * Minimized energy: 1710.352516412735
* Info * Saved coordinates for step 29400
* Info * Minimized energy: 1684.4164389371872
* Info * Saved coordinates for step 29600
* Info * Minimized energy: 1704.6233137845993
* Info * Saved coordinates for step 29800
* Info * Minimized energy: 1703.82393181324
* Info * Saved coordinates for step 30000
* Info * Minimized energy: 1672.3961992263794
* Info * Saved coordinates for step 30200
* Info * Minimized energy: 1672.5638521909714
* Info * Saved coordinates for step 30400
* Info * Minimized energy: 1655.080955862999
* Info * Saved coordinates for step 30600
* Info * Minimized energy: 1671.6834859848022
* Info * Saved coordinates for step 30800
* Info * Minimized energy: 1673.6869477629662
* Info * Saved coordinates for step 31000
* Info * Minimized energy: 1657.6211230754852
* Info * Saved coordinates for step 31200
* Info * Minimized energy: 1680.9596975147724
* Info * Saved coordinates for step 31400
* Info * Minimized energy: 1708.3319111913443
* Info * Saved coordinates for step 31600
* Info * Minimized energy: 1680.333521604538
* Info * Saved coordinates for step 31800
* Info * Minimized energy: 1733.5229585766792
* Info * Saved coordinates for step 32000
* Info * Minimized energy: 1678.2798300981522
* Info * Saved coordinates for step 32200
* Info * Minimized energy: 1683.9787719249725
* Info * Saved coordinates for step 32400
* Info * Minimized energy: 1728.430027604103
* Info * Saved coordinates for step 32600
* Info * Minimized energy: 1630.494354724884
* Info * Saved coordinates for step 32800
* Info * Minimized energy: 1660.211130976677
* Info * Saved coordinates for step 33000
* Info * Minimized energy: 1655.7404009103775
* Info * Saved coordinates for step 33200
* Info * Minimized energy: 1653.5492098927498
* Info * Saved coordinates for step 33400
* Info * Minimized energy: 1692.983805179596
* Info * Saved coordinates for step 33600
* Info * Minimized energy: 1702.2086563110352
* Info * Saved coordinates for step 33800
* Info * Minimized energy: 1685.7653579115868
* Info * Saved coordinates for step 34000
* Info * Minimized energy: 1671.413040280342
* Info * Saved coordinates for step 34200
* Info * Minimized energy: 1680.5441534221172
* Info * Saved coordinates for step 34400
* Info * Minimized energy: 1686.0361524522305
* Info * Saved coordinates for step 34600
* Info * Minimized energy: 1672.0484097898006
* Info * Saved coordinates for step 34800
* Info * Minimized energy: 1655.7319561839104
* Info * Saved coordinates for step 35000
* Info * Minimized energy: 1670.5675796568394
* Info * Saved coordinates for step 35200
* Info * Minimized energy: 1660.2359159588814
* Info * Saved coordinates for step 35400
* Info * Minimized energy: 1723.8239603042603
* Info * Saved coordinates for step 35600
* Info * Minimized energy: 1669.1979645490646
* Info * Saved coordinates for step 35800
* Info * Minimized energy: 1653.150118291378
* Info * Saved coordinates for step 36000
* Info * Minimized energy: 1689.8258673846722
* Info * Saved coordinates for step 36200
* Info * Minimized energy: 1686.0043043494225
* Info * Saved coordinates for step 36400
* Info * Minimized energy: 1692.2212767004967
* Info * Saved coordinates for step 36600
* Info * Minimized energy: 1708.236263513565
* Info * Saved coordinates for step 36800
* Info * Minimized energy: 1678.9968178868294
* Info * Saved coordinates for step 37000
* Info * Minimized energy: 1666.8676787018776
* Info * Saved coordinates for step 37200
* Info * Minimized energy: 1657.0669391155243
* Info * Saved coordinates for step 37400
* Info * Minimized energy: 1677.511938393116
* Info * Saved coordinates for step 37600
* Info * Minimized energy: 1665.2867010831833
* Info * Saved coordinates for step 37800
* Info * Minimized energy: 1657.7656290829182
* Info * Saved coordinates for step 38000
* Info * Minimized energy: 1663.5209134817123
* Info * Saved coordinates for step 38200
* Info * Minimized energy: 1663.9222984313965
* Info * Saved coordinates for step 38400
* Info * Minimized energy: 1658.4751383662224
* Info * Saved coordinates for step 38600
* Info * Minimized energy: 1666.3533607721329
* Info * Saved coordinates for step 38800
* Info * Minimized energy: 1680.4991365671158
* Info * Saved coordinates for step 39000
* Info * Minimized energy: 1678.369568169117
* Info * Saved coordinates for step 39200
* Info * Minimized energy: 1684.078638970852
* Info * Saved coordinates for step 39400
* Info * Minimized energy: 1670.2707547545433
* Info * Saved coordinates for step 39600
* Info * Minimized energy: 1695.1017092466354
* Info * Saved coordinates for step 39800
* Info * Minimized energy: 1687.9714651107788
* Info * Saved coordinates for step 40000
* Info * Minimized energy: 1666.5032325387
* Info * Saved coordinates for step 40200
* Info * Minimized energy: 1678.2060804963112
* Info * Saved coordinates for step 40400
* Info * Minimized energy: 1666.3618072867393
* Info * Saved coordinates for step 40600
* Info * Minimized energy: 1684.0858156085014
* Info * Saved coordinates for step 40800
* Info * Minimized energy: 1666.0635766983032
* Info * Saved coordinates for step 41000
* Info * Minimized energy: 1675.5248917341232
* Info * Saved coordinates for step 41200
* Info * Minimized energy: 1699.2471492290497
* Info * Saved coordinates for step 41400
* Info * Minimized energy: 1692.0617645978928
* Info * Saved coordinates for step 41600
* Info * Minimized energy: 1682.5613943338394
* Info * Saved coordinates for step 41800
* Info * Minimized energy: 1683.176533550024
* Info * Saved coordinates for step 42000
* Info * Minimized energy: 1663.3696638345718
* Info * Saved coordinates for step 42200
* Info * Minimized energy: 1666.0758202075958
* Info * Saved coordinates for step 42400
* Info * Minimized energy: 1698.0822186470032
* Info * Saved coordinates for step 42600
* Info * Minimized energy: 1682.4726001024246
* Info * Saved coordinates for step 42800
* Info * Minimized energy: 1642.9378886818886
* Info * Saved coordinates for step 43000
* Info * Minimized energy: 1640.0848786830902
* Info * Saved coordinates for step 43200
* Info * Minimized energy: 1638.131539195776
* Info * Saved coordinates for step 43400
* Info * Minimized energy: 1646.3154257535934
* Info * Saved coordinates for step 43600
* Info * Minimized energy: 1683.5731253027916
* Info * Saved coordinates for step 43800
* Info * Minimized energy: 1650.0796197652817
* Info * Saved coordinates for step 44000
* Info * Minimized energy: 1630.9225362539291
* Info * Saved coordinates for step 44200
* Info * Minimized energy: 1650.1533258259296
* Info * Saved coordinates for step 44400
* Info * Minimized energy: 1617.8280853033066
* Info * Saved coordinates for step 44600
* Info * Minimized energy: 1629.568809747696
* Info * Saved coordinates for step 44800
* Info * Minimized energy: 1643.6250585317612
* Info * Saved coordinates for step 45000
* Info * Minimized energy: 1623.6018466949463
* Info * Saved coordinates for step 45200
* Info * Minimized energy: 1636.3413794636726
* Info * Saved coordinates for step 45400
* Info * Minimized energy: 1654.1841477751732
* Info * Saved coordinates for step 45600
* Info * Minimized energy: 1642.5187499523163
* Info * Saved coordinates for step 45800
* Info * Minimized energy: 1649.3668453097343
* Info * Saved coordinates for step 46000
* Info * Minimized energy: 1667.6458382606506
* Info * Saved coordinates for step 46200
* Info * Minimized energy: 1653.4894766807556
* Info * Saved coordinates for step 46400
* Info * Minimized energy: 1662.3728858232498
* Info * Saved coordinates for step 46600
* Info * Minimized energy: 1645.778172135353
* Info * Saved coordinates for step 46800
* Info * Minimized energy: 1659.5529046058655
* Info * Saved coordinates for step 47000
* Info * Minimized energy: 1692.9702701866627
* Info * Saved coordinates for step 47200
* Info * Minimized energy: 1632.9286402463913
* Info * Saved coordinates for step 47400
* Info * Minimized energy: 1654.717675626278
* Info * Saved coordinates for step 47600
* Info * Minimized energy: 1657.8518676757812
* Info * Saved coordinates for step 47800
* Info * Minimized energy: 1656.251745223999
* Info * Saved coordinates for step 48000
* Info * Minimized energy: 1658.9721381664276
* Info * Saved coordinates for step 48200
* Info * Minimized energy: 1679.4698075652122
* Info * Saved coordinates for step 48400
* Info * Minimized energy: 1650.6787678003311
* Info * Saved coordinates for step 48600
* Info * Minimized energy: 1668.7670662403107
* Info * Saved coordinates for step 48800
* Info * Minimized energy: 1638.2412309646606
* Info * Saved coordinates for step 49000
* Info * Minimized energy: 1658.3208650946617
* Info * Saved coordinates for step 49200
* Info * Minimized energy: 1654.333293557167
* Info * Saved coordinates for step 49400
* Info * Minimized energy: 1643.610674381256
* Info * Saved coordinates for step 49600
* Info * Minimized energy: 1646.6410180330276
* Info * Saved coordinates for step 49800
* Info * Minimized energy: 1650.7398526668549
* Info * Saved coordinates for step 50000
* Info * Minimized energy: 1647.978950381279
* Info * Saved coordinates for step 50200
* Info * Minimized energy: 1709.6216312646866
* Info * Saved coordinates for step 50400
* Info * Minimized energy: 1712.187863945961
* Info * Saved coordinates for step 50600
* Info * Minimized energy: 1690.6738731861115
* Info * Saved coordinates for step 50800
* Info * Minimized energy: 1716.4765757322311
* Info * Saved coordinates for step 51000
* Info * Minimized energy: 1745.1871750354767
* Info * Saved coordinates for step 51200
* Info * Minimized energy: 1712.5074628591537
* Info * Saved coordinates for step 51400
* Info * Minimized energy: 1723.1986060142517
* Info * Saved coordinates for step 51600
* Info * Minimized energy: 1688.9645961523056
* Info * Saved coordinates for step 51800
* Info * Minimized energy: 1738.6815441846848
* Info * Saved coordinates for step 52000
* Info * Minimized energy: 1695.2964222431183
* Info * Saved coordinates for step 52200
* Info * Minimized energy: 1653.1344903707504
* Info * Saved coordinates for step 52400
* Info * Minimized energy: 1672.3849534392357
* Info * Saved coordinates for step 52600
* Info * Minimized energy: 1666.3897224068642
* Info * Saved coordinates for step 52800
* Info * Minimized energy: 1698.930967450142
* Info * Saved coordinates for step 53000
* Info * Minimized energy: 1702.4923985004425
* Info * Saved coordinates for step 53200
* Info * Minimized energy: 1678.089069724083
* Info * Saved coordinates for step 53400
* Info * Minimized energy: 1691.1692249774933
* Info * Saved coordinates for step 53600
* Info * Minimized energy: 1707.4337458610535
* Info * Saved coordinates for step 53800
* Info * Minimized energy: 1686.3831732273102
* Info * Saved coordinates for step 54000
* Info * Minimized energy: 1670.331062555313
* Info * Saved coordinates for step 54200
* Info * Minimized energy: 1692.0971705317497
* Info * Saved coordinates for step 54400
* Info * Minimized energy: 1696.6222566366196
* Info * Saved coordinates for step 54600
* Info * Minimized energy: 1701.3831726312637
* Info * Saved coordinates for step 54800
* Info * Minimized energy: 1692.1930838823318
* Info * Saved coordinates for step 55000
* Info * Minimized energy: 1673.0574429035187
* Info * Saved coordinates for step 55200
* Info * Minimized energy: 1670.1504077911377
* Info * Saved coordinates for step 55400
* Info * Minimized energy: 1678.1820005178452
* Info * Saved coordinates for step 55600
* Info * Minimized energy: 1717.2771507799625
* Info * Saved coordinates for step 55800
* Info * Minimized energy: 1693.216609299183
* Info * Saved coordinates for step 56000
* Info * Minimized energy: 1706.5884444713593
* Info * Saved coordinates for step 56200
* Info * Minimized energy: 1691.1827552318573
* Info * Saved coordinates for step 56400
* Info * Minimized energy: 1726.0391075611115
* Info * Saved coordinates for step 56600
* Info * Minimized energy: 1712.8682088255882
* Info * Saved coordinates for step 56800
* Info * Minimized energy: 1707.8546063899994
* Info * Saved coordinates for step 57000
* Info * Minimized energy: 1705.0674893558025
* Info * Saved coordinates for step 57200
* Info * Minimized energy: 1691.9576062560081
* Info * Saved coordinates for step 57400
* Info * Minimized energy: 1694.3276310563087
* Info * Saved coordinates for step 57600
* Info * Minimized energy: 1701.7856097519398
* Info * Saved coordinates for step 57800
* Info * Minimized energy: 1645.0386717617512
* Info * Saved coordinates for step 58000
* Info * Minimized energy: 1653.7510392069817
* Info * Saved coordinates for step 58200
* Info * Minimized energy: 1683.166575178504
* Info * Saved coordinates for step 58400
* Info * Minimized energy: 1697.4435250759125
* Info * Saved coordinates for step 58600
* Info * Minimized energy: 1726.5510849356651
* Info * Saved coordinates for step 58800
* Info * Minimized energy: 1710.4438603520393
* Info * Saved coordinates for step 59000
* Info * Minimized energy: 1665.7730065584183
* Info * Saved coordinates for step 59200
* Info * Minimized energy: 1678.1762709021568
* Info * Saved coordinates for step 59400
* Info * Minimized energy: 1651.9790868759155
* Info * Saved coordinates for step 59600
* Info * Minimized energy: 1687.0441878437996
* Info * Saved coordinates for step 59800
* Info * Minimized energy: 1695.8435726165771
* Info * Saved coordinates for step 60000
* Info * Minimized energy: 1641.8917416334152
* Info * Saved coordinates for step 60200
* Info * Minimized energy: 1699.165721833706
* Info * Saved coordinates for step 60400
* Info * Minimized energy: 1668.26692289114
* Info * Saved coordinates for step 60600
* Info * Minimized energy: 1685.7870880365372
* Info * Saved coordinates for step 60800
* Info * Minimized energy: 1681.0899341106415
* Info * Saved coordinates for step 61000
* Info * Minimized energy: 1701.3276235163212
* Info * Saved coordinates for step 61200
* Info * Minimized energy: 1682.7089893519878
* Info * Saved coordinates for step 61400
* Info * Minimized energy: 1658.082983583212
* Info * Saved coordinates for step 61600
* Info * Minimized energy: 1665.607833981514
* Info * Saved coordinates for step 61800
* Info * Minimized energy: 1680.2788616418839
* Info * Saved coordinates for step 62000
* Info * Minimized energy: 1701.7502372860909
* Info * Saved coordinates for step 62200
* Info * Minimized energy: 1690.5811036229134
* Info * Saved coordinates for step 62400
* Info * Minimized energy: 1663.856436431408
* Info * Saved coordinates for step 62600
* Info * Minimized energy: 1682.8883793354034
* Info * Saved coordinates for step 62800
* Info * Minimized energy: 1741.9509363174438
* Info * Saved coordinates for step 63000
* Info * Minimized energy: 1728.8235076665878
* Info * Saved coordinates for step 63200
* Info * Minimized energy: 1691.1646778583527
* Info * Saved coordinates for step 63400
* Info * Minimized energy: 1686.8051154613495
* Info * Saved coordinates for step 63600
* Info * Minimized energy: 1715.0675943493843
* Info * Saved coordinates for step 63800
* Info * Minimized energy: 1710.8993206620216
* Info * Saved coordinates for step 64000
* Info * Minimized energy: 1687.3217507600784
* Info * Saved coordinates for step 64200
* Info * Minimized energy: 1665.7313215732574
* Info * Saved coordinates for step 64400
* Info * Minimized energy: 1664.6988290548325
* Info * Saved coordinates for step 64600
* Info * Minimized energy: 1683.303721189499
* Info * Saved coordinates for step 64800
* Info * Minimized energy: 1715.1847383379936
* Info * Saved coordinates for step 65000
* Info * Minimized energy: 1734.5005425214767
* Info * Saved coordinates for step 65200
* Info * Minimized energy: 1691.6983513832092
* Info * Saved coordinates for step 65400
* Info * Minimized energy: 1704.2380969524384
* Info * Saved coordinates for step 65600
* Info * Minimized energy: 1708.7483266592026
* Info * Saved coordinates for step 65800
* Info * Minimized energy: 1718.9132087230682
* Info * Saved coordinates for step 66000
* Info * Minimized energy: 1707.1023661494255
* Info * Saved coordinates for step 66200
* Info * Minimized energy: 1713.0191550850868
* Info * Saved coordinates for step 66400
* Info * Minimized energy: 1739.7075060009956
* Info * Saved coordinates for step 66600
* Info * Minimized energy: 1719.8601301312447
* Info * Saved coordinates for step 66800
* Info * Minimized energy: 1721.3559865355492
* Info * Saved coordinates for step 67000
* Info * Minimized energy: 1696.3164556324482
* Info * Saved coordinates for step 67200
* Info * Minimized energy: 1713.703111410141
* Info * Saved coordinates for step 67400
* Info * Minimized energy: 1705.9688947498798
* Info * Saved coordinates for step 67600
* Info * Minimized energy: 1729.2555828094482
* Info * Saved coordinates for step 67800
* Info * Minimized energy: 1670.4268616139889
* Info * Saved coordinates for step 68000
* Info * Minimized energy: 1707.439880847931
* Info * Saved coordinates for step 68200
* Info * Minimized energy: 1731.1564534902573
* Info * Saved coordinates for step 68400
* Info * Minimized energy: 1686.220472857356
* Info * Saved coordinates for step 68600
* Info * Minimized energy: 1693.588491678238
* Info * Saved coordinates for step 68800
* Info * Minimized energy: 1675.0731472969055
* Info * Saved coordinates for step 69000
* Info * Minimized energy: 1673.4902813136578
* Info * Saved coordinates for step 69200
* Info * Minimized energy: 1667.7772098183632
* Info * Saved coordinates for step 69400
* Info * Minimized energy: 1680.0753020048141
* Info * Saved coordinates for step 69600
* Info * Minimized energy: 1696.4744671583176
* Info * Saved coordinates for step 69800
* Info * Minimized energy: 1676.1965032815933
* Info * Saved coordinates for step 70000
* Info * Minimized energy: 1687.466824710369
* Info * Saved coordinates for step 70200
* Info * Minimized energy: 1689.9222331047058
* Info * Saved coordinates for step 70400
* Info * Minimized energy: 1680.8254896998405
* Info * Saved coordinates for step 70600
* Info * Minimized energy: 1703.7046815752983
* Info * Saved coordinates for step 70800
* Info * Minimized energy: 1706.0568584799767
* Info * Saved coordinates for step 71000
* Info * Minimized energy: 1695.6557171940804
* Info * Saved coordinates for step 71200
* Info * Minimized energy: 1745.3437408208847
* Info * Saved coordinates for step 71400
* Info * Minimized energy: 1727.6781095266342
* Info * Saved coordinates for step 71600
* Info * Minimized energy: 1742.4006510972977
* Info * Saved coordinates for step 71800
* Info * Minimized energy: 1722.131737112999
* Info * Saved coordinates for step 72000
* Info * Minimized energy: 1717.8382652401924
* Info * Saved coordinates for step 72200
* Info * Minimized energy: 1717.744766175747
* Info * Saved coordinates for step 72400
* Info * Minimized energy: 1706.832837343216
* Info * Saved coordinates for step 72600
* Info * Minimized energy: 1718.1892884969711
* Info * Saved coordinates for step 72800
* Info * Minimized energy: 1704.5090956687927
* Info * Saved coordinates for step 73000
* Info * Minimized energy: 1684.3724479675293
* Info * Saved coordinates for step 73200
* Info * Minimized energy: 1717.8428531885147
* Info * Saved coordinates for step 73400
* Info * Minimized energy: 1704.3052150011063
* Info * Saved coordinates for step 73600
* Info * Minimized energy: 1694.5071040391922
* Info * Saved coordinates for step 73800
* Info * Minimized energy: 1695.3418064117432
* Info * Saved coordinates for step 74000
* Info * Minimized energy: 1699.0311195850372
* Info * Saved coordinates for step 74200
* Info * Minimized energy: 1705.6895664930344
* Info * Saved coordinates for step 74400
* Info * Minimized energy: 1716.8089921474457
* Info * Saved coordinates for step 74600
* Info * Minimized energy: 1733.935974240303
* Info * Saved coordinates for step 74800
* Info * Minimized energy: 1698.8946422338486
* Info * Saved coordinates for step 75000
* Info * Minimized energy: 1717.2951312065125
* Info * Saved coordinates for step 75200
* Info * Minimized energy: 1708.6237351298332
* Info * Saved coordinates for step 75400
* Info * Minimized energy: 1698.0268615484238
* Info * Saved coordinates for step 75600
* Info * Minimized energy: 1677.8882088661194
* Info * Saved coordinates for step 75800
* Info * Minimized energy: 1686.5271928310394
* Info * Saved coordinates for step 76000
* Info * Minimized energy: 1704.1719794273376
* Info * Saved coordinates for step 76200
* Info * Minimized energy: 1665.681584596634
* Info * Saved coordinates for step 76400
* Info * Minimized energy: 1646.354943871498
* Info * Saved coordinates for step 76600
* Info * Minimized energy: 1709.1004288196564
* Info * Saved coordinates for step 76800
* Info * Minimized energy: 1669.4106159210205
* Info * Saved coordinates for step 77000
* Info * Minimized energy: 1754.5406749248505
* Info * Saved coordinates for step 77200
* Info * Minimized energy: 1721.0510711669922
* Info * Saved coordinates for step 77400
* Info * Minimized energy: 1702.2043297290802
* Info * Saved coordinates for step 77600
* Info * Minimized energy: 1739.7059659957886
* Info * Saved coordinates for step 77800
* Info * Minimized energy: 1664.4810975790024
* Info * Saved coordinates for step 78000
* Info * Minimized energy: 1753.9405966997147
* Info * Saved coordinates for step 78200
* Info * Minimized energy: 1659.7301692962646
* Info * Saved coordinates for step 78400
* Info * Minimized energy: 1632.7337990999222
* Info * Saved coordinates for step 78600
* Info * Minimized energy: 1703.8913288116455
* Info * Saved coordinates for step 78800
* Info * Minimized energy: 1651.7058806419373
* Info * Saved coordinates for step 79000
* Info * Minimized energy: 1676.7754808664322
* Info * Saved coordinates for step 79200
* Info * Minimized energy: 1668.5427815914154
* Info * Saved coordinates for step 79400
* Info * Minimized energy: 1689.6001472473145
* Info * Saved coordinates for step 79600
* Info * Minimized energy: 1681.2646144628525
* Info * Saved coordinates for step 79800
* Info * Minimized energy: 1683.5950957536697
* Info * Saved coordinates for step 80000
* Info * Minimized energy: 1682.5235403776169
* Info * Saved coordinates for step 80200
* Info * Minimized energy: 1680.007816195488
* Info * Saved coordinates for step 80400
* Info * Minimized energy: 1684.460402727127
* Info * Saved coordinates for step 80600
* Info * Minimized energy: 1660.1310704946518
* Info * Saved coordinates for step 80800
* Info * Minimized energy: 1675.231297492981
* Info * Saved coordinates for step 81000
* Info * Minimized energy: 1674.203895419836
* Info * Saved coordinates for step 81200
* Info * Minimized energy: 1692.0206569433212
* Info * Saved coordinates for step 81400
* Info * Minimized energy: 1708.3330756425858
* Info * Saved coordinates for step 81600
* Info * Minimized energy: 1652.8553906679153
* Info * Saved coordinates for step 81800
* Info * Minimized energy: 1656.8788691163063
* Info * Saved coordinates for step 82000
* Info * Minimized energy: 1673.545471072197
* Info * Saved coordinates for step 82200
* Info * Minimized energy: 1701.5549750328064
* Info * Saved coordinates for step 82400
* Info * Minimized energy: 1699.5102177262306
* Info * Saved coordinates for step 82600
* Info * Minimized energy: 1719.955133229494
* Info * Saved coordinates for step 82800
* Info * Minimized energy: 1711.7322177886963
* Info * Saved coordinates for step 83000
* Info * Minimized energy: 1726.0852012634277
* Info * Saved coordinates for step 83200
* Info * Minimized energy: 1716.3183867931366
* Info * Saved coordinates for step 83400
* Info * Minimized energy: 1711.3620874881744
* Info * Saved coordinates for step 83600
* Info * Minimized energy: 1712.650275349617
* Info * Saved coordinates for step 83800
* Info * Minimized energy: 1749.8768587112427
* Info * Saved coordinates for step 84000
* Info * Minimized energy: 1728.8311705589294
* Info * Saved coordinates for step 84200
* Info * Minimized energy: 1725.978437423706
* Info * Saved coordinates for step 84400
* Info * Minimized energy: 1779.0787303447723
* Info * Saved coordinates for step 84600
* Info * Minimized energy: 1709.6432332992554
* Info * Saved coordinates for step 84800
* Info * Minimized energy: 1720.4568427801132
* Info * Saved coordinates for step 85000
* Info * Minimized energy: 1713.0995526313782
* Info * Saved coordinates for step 85200
* Info * Minimized energy: 1719.486818432808
* Info * Saved coordinates for step 85400
* Info * Minimized energy: 1727.5082396268845
* Info * Saved coordinates for step 85600
* Info * Minimized energy: 1707.6499055624008
* Info * Saved coordinates for step 85800
* Info * Minimized energy: 1712.8581229448318
* Info * Saved coordinates for step 86000
* Info * Minimized energy: 1708.6678696870804
* Info * Saved coordinates for step 86200
* Info * Minimized energy: 1691.3191356658936
* Info * Saved coordinates for step 86400
* Info * Minimized energy: 1714.9747726917267
* Info * Saved coordinates for step 86600
* Info * Minimized energy: 1720.8672989606857
* Info * Saved coordinates for step 86800
* Info * Minimized energy: 1724.1456953287125
* Info * Saved coordinates for step 87000
* Info * Minimized energy: 1747.7379312515259
* Info * Saved coordinates for step 87200
* Info * Minimized energy: 1733.2876340150833
* Info * Saved coordinates for step 87400
* Info * Minimized energy: 1711.2574661970139
* Info * Saved coordinates for step 87600
* Info * Minimized energy: 1736.1333285570145
* Info * Saved coordinates for step 87800
* Info * Minimized energy: 1693.7807918787003
* Info * Saved coordinates for step 88000
* Info * Minimized energy: 1724.0940126180649
* Info * Saved coordinates for step 88200
* Info * Minimized energy: 1705.1494755744934
* Info * Saved coordinates for step 88400
* Info * Minimized energy: 1728.0138986110687
* Info * Saved coordinates for step 88600
* Info * Minimized energy: 1749.4708530902863
* Info * Saved coordinates for step 88800
* Info * Minimized energy: 1736.6900829076767
* Info * Saved coordinates for step 89000
* Info * Minimized energy: 1723.7890434265137
* Info * Saved coordinates for step 89200
* Info * Minimized energy: 1710.9641189575195
* Info * Saved coordinates for step 89400
* Info * Minimized energy: 1702.691679239273
* Info * Saved coordinates for step 89600
* Info * Minimized energy: 1719.8177967071533
* Info * Saved coordinates for step 89800
* Info * Minimized energy: 1714.6431338787079
* Info * Saved coordinates for step 90000
* Info * Minimized energy: 1690.1494491100311
* Info * Saved coordinates for step 90200
* Info * Minimized energy: 1702.5580883026123
* Info * Saved coordinates for step 90400
* Info * Minimized energy: 1674.54716527462
* Info * Saved coordinates for step 90600
* Info * Minimized energy: 1673.7365691661835
* Info * Saved coordinates for step 90800
* Info * Minimized energy: 1654.8971028327942
* Info * Saved coordinates for step 91000
* Info * Minimized energy: 1695.2111101150513
* Info * Saved coordinates for step 91200
* Info * Minimized energy: 1657.435625076294
* Info * Saved coordinates for step 91400
* Info * Minimized energy: 1627.7293713092804
* Info * Saved coordinates for step 91600
* Info * Minimized energy: 1639.3391128778458
* Info * Saved coordinates for step 91800
* Info * Minimized energy: 1626.9777643680573
* Info * Saved coordinates for step 92000
* Info * Minimized energy: 1635.1128342151642
* Info * Saved coordinates for step 92200
* Info * Minimized energy: 1665.774218082428
* Info * Saved coordinates for step 92400
* Info * Minimized energy: 1667.8745412826538
* Info * Saved coordinates for step 92600
* Info * Minimized energy: 1668.2799968719482
* Info * Saved coordinates for step 92800
* Info * Minimized energy: 1700.162091255188
* Info * Saved coordinates for step 93000
* Info * Minimized energy: 1696.0910186767578
* Info * Saved coordinates for step 93200
* Info * Minimized energy: 1749.290964603424
* Info * Saved coordinates for step 93400
* Info * Minimized energy: 1750.9580578804016
* Info * Saved coordinates for step 93600
* Info * Minimized energy: 1725.7984218597412
* Info * Saved coordinates for step 93800
* Info * Minimized energy: 1751.7751669883728
* Info * Saved coordinates for step 94000
* Info * Minimized energy: 1744.1482157707214
* Info * Saved coordinates for step 94200
* Info * Minimized energy: 1731.8847103118896
* Info * Saved coordinates for step 94400
* Info * Minimized energy: 1702.8869805335999
* Info * Saved coordinates for step 94600
* Info * Minimized energy: 1763.2389113903046
* Info * Saved coordinates for step 94800
* Info * Minimized energy: 1727.723908662796
* Info * Saved coordinates for step 95000
* Info * Minimized energy: 1691.261953830719
* Info * Saved coordinates for step 95200
* Info * Minimized energy: 1672.315006017685
* Info * Saved coordinates for step 95400
* Info * Minimized energy: 1713.9877662658691
* Info * Saved coordinates for step 95600
* Info * Minimized energy: 1707.4733488559723
* Info * Saved coordinates for step 95800
* Info * Minimized energy: 1653.8759047985077
* Info * Saved coordinates for step 96000
* Info * Minimized energy: 1675.769744515419
* Info * Saved coordinates for step 96200
* Info * Minimized energy: 1705.002292394638
* Info * Saved coordinates for step 96400
* Info * Minimized energy: 1729.2445976734161
* Info * Saved coordinates for step 96600
* Info * Minimized energy: 1737.3967673778534
* Info * Saved coordinates for step 96800
* Info * Minimized energy: 1734.4447484016418
* Info * Saved coordinates for step 97000
* Info * Minimized energy: 1725.2822077274323
* Info * Saved coordinates for step 97200
* Info * Minimized energy: 1734.3673367500305
* Info * Saved coordinates for step 97400
* Info * Minimized energy: 1697.922344326973
* Info * Saved coordinates for step 97600
* Info * Minimized energy: 1709.5104469060898
* Info * Saved coordinates for step 97800
* Info * Minimized energy: 1720.753447175026
* Info * Saved coordinates for step 98000
* Info * Minimized energy: 1722.7471588850021
* Info * Saved coordinates for step 98200
* Info * Minimized energy: 1708.7501982450485
* Info * Saved coordinates for step 98400
* Info * Minimized energy: 1724.4080095291138
* Info * Saved coordinates for step 98600
* Info * Minimized energy: 1734.121146082878
* Info * Saved coordinates for step 98800
* Info * Minimized energy: 1777.6181681156158
* Info * Saved coordinates for step 99000
* Info * Minimized energy: 1760.337054014206
* Info * Saved coordinates for step 99200
* Info * Minimized energy: 1752.6971142292023
* Info * Saved coordinates for step 99400
* Info * Minimized energy: 1744.8232548236847
* Info * Saved coordinates for step 99600
* Info * Minimized energy: 1706.1954275369644
* Info * Saved coordinates for step 99800
* Info * Minimized energy: 1695.461933016777
* Info * Saved coordinates for step 100000
* Info * Conformational sampling completed!
* Info * Number of conformations: 500
* Info * Filtering for unique conformers
* Info *
Number of unique conformers: 485
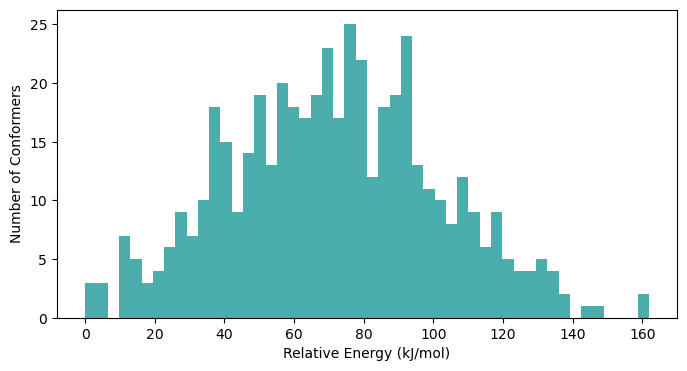
# plot the distribution of the relative energies of the conformers
import matplotlib.pyplot as plt
import numpy as np
fig, ax = plt.subplots(figsize=(8, 4))
ax.hist(conformers_dict['energies']-np.min(conformers_dict['energies']),
bins=50,
color='darkcyan',
alpha=0.7)
plt.xlabel('Relative Energy (kJ/mol)')
plt.ylabel('Number of Conformers')
plt.show()
opm_dyn.show_conformers(number=3)* Info *
Conformation 1: Energy: 1617.202 kJ/mol, Weight: 0.3124
* Info *
Conformation 2: Energy: 1617.256 kJ/mol, Weight: 0.3057
* Info *
Conformation 3: Energy: 1617.828 kJ/mol, Weight: 0.2431
# Write it to an XYZ file
conformers_dict['molecules'][0].write_xyz_file('tq-polymer-0.xyz')Conformational search using MD for multiple molecules¶
When dealing with multiple molecules simultaneously, VeloxChem supports a multi-molecule conformational search. This approach automatically generates force fields for each molecule and employs the OpenMMDynamics driver with a centroid restraint force to prevent molecules from drifting apart during a high-temperature MD simulation. A user-defined number of snapshots are then energy-minimized to yield a representative set of low-energy conformers.
Load your molecules and create a list containing those molecules.
mol1 = vlx.Molecule.read_smiles('C([C@@H]1[C@H]([C@@H]([C@H](C(O1)O)O)O)O)O')
mol2 = vlx.Molecule.read_smiles('O')
mol3 = vlx.Molecule.read_smiles('O')
molecules = [mol1, mol2, mol3]Initiate and run the conformational search for multiple molecules.
omm = vlx.OpenMMDynamics()
conformer_dict = omm.conformational_sampling_multiple(molecules,
temperature = 800,
nsteps = 100000,
snapshots = 40,
lowest_conformations = 3)* Info * Generating system...
* Info * Water molecule detected. Using tip3p water model.
***********
* Warning * CONECT records not found in the PDB file.The connectivity matrix will be used to determine the bonds.
***********
* Info * Unique Residues: [('M01', 1), ('M02', 2), ('M03', 3)], saved as molecules.
* Info * PBC detected from the PDB file.
* Info * Added XML Files: ['molecule_1.xml', 'molecule_2.xml']
* Info * Applying centroid bond force to the system...
* Info * Running high-temperature MD for 0.20 ns...
* Info * Saved coordinates for step 2500
* Info * Energy for conformer 1/40: 395.1134 kJ/mol
* Info * Saved coordinates for step 5000
* Info * Energy for conformer 2/40: 352.5593 kJ/mol
* Info * Saved coordinates for step 7500
* Info * Energy for conformer 3/40: 437.1005 kJ/mol
* Info * Saved coordinates for step 10000
* Info * Energy for conformer 4/40: 375.8574 kJ/mol
* Info * Saved coordinates for step 12500
* Info * Energy for conformer 5/40: 427.6551 kJ/mol
* Info * Saved coordinates for step 15000
* Info * Energy for conformer 6/40: 424.2773 kJ/mol
* Info * Saved coordinates for step 17500
* Info * Energy for conformer 7/40: 377.2951 kJ/mol
* Info * Saved coordinates for step 20000
* Info * Energy for conformer 8/40: 423.5491 kJ/mol
* Info * Saved coordinates for step 22500
* Info * Energy for conformer 9/40: 420.5697 kJ/mol
* Info * Saved coordinates for step 25000
* Info * Energy for conformer 10/40: 388.6808 kJ/mol
* Info * Saved coordinates for step 27500
* Info * Energy for conformer 11/40: 378.8103 kJ/mol
* Info * Saved coordinates for step 30000
* Info * Energy for conformer 12/40: 460.0286 kJ/mol
* Info * Saved coordinates for step 32500
* Info * Energy for conformer 13/40: 462.8509 kJ/mol
* Info * Saved coordinates for step 35000
* Info * Energy for conformer 14/40: 364.1092 kJ/mol
* Info * Saved coordinates for step 37500
* Info * Energy for conformer 15/40: 431.8992 kJ/mol
* Info * Saved coordinates for step 40000
* Info * Energy for conformer 16/40: 317.3371 kJ/mol
* Info * Saved coordinates for step 42500
* Info * Energy for conformer 17/40: 402.2867 kJ/mol
* Info * Saved coordinates for step 45000
* Info * Energy for conformer 18/40: 430.9367 kJ/mol
* Info * Saved coordinates for step 47500
* Info * Energy for conformer 19/40: 384.4058 kJ/mol
* Info * Saved coordinates for step 50000
* Info * Energy for conformer 20/40: 454.3422 kJ/mol
* Info * Saved coordinates for step 52500
* Info * Energy for conformer 21/40: 376.1228 kJ/mol
* Info * Saved coordinates for step 55000
* Info * Energy for conformer 22/40: 395.2607 kJ/mol
* Info * Saved coordinates for step 57500
* Info * Energy for conformer 23/40: 304.7673 kJ/mol
* Info * Saved coordinates for step 60000
* Info * Energy for conformer 24/40: 402.8433 kJ/mol
* Info * Saved coordinates for step 62500
* Info * Energy for conformer 25/40: 389.3687 kJ/mol
* Info * Saved coordinates for step 65000
* Info * Energy for conformer 26/40: 380.6306 kJ/mol
* Info * Saved coordinates for step 67500
* Info * Energy for conformer 27/40: 365.7653 kJ/mol
* Info * Saved coordinates for step 70000
* Info * Energy for conformer 28/40: 399.0216 kJ/mol
* Info * Saved coordinates for step 72500
* Info * Energy for conformer 29/40: 411.8805 kJ/mol
* Info * Saved coordinates for step 75000
* Info * Energy for conformer 30/40: 361.8857 kJ/mol
* Info * Saved coordinates for step 77500
* Info * Energy for conformer 31/40: 411.2035 kJ/mol
* Info * Saved coordinates for step 80000
* Info * Energy for conformer 32/40: 344.5685 kJ/mol
* Info * Saved coordinates for step 82500
* Info * Energy for conformer 33/40: 393.0867 kJ/mol
* Info * Saved coordinates for step 85000
* Info * Energy for conformer 34/40: 426.2565 kJ/mol
* Info * Saved coordinates for step 87500
* Info * Energy for conformer 35/40: 388.5971 kJ/mol
* Info * Saved coordinates for step 90000
* Info * Energy for conformer 36/40: 475.6388 kJ/mol
* Info * Saved coordinates for step 92500
* Info * Energy for conformer 37/40: 430.8434 kJ/mol
* Info * Saved coordinates for step 95000
* Info * Energy for conformer 38/40: 359.4068 kJ/mol
* Info * Saved coordinates for step 97500
* Info * Energy for conformer 39/40: 367.8442 kJ/mol
* Info * Saved coordinates for step 100000
* Info * Energy for conformer 40/40: 386.6349 kJ/mol
* Info * Recalculating energies for the conformations...
* Info * Minimized energy of conformer 0: 158.0144 kJ/mol
* Info * Minimized energy of conformer 1: 140.4215 kJ/mol
* Info * Minimized energy of conformer 2: 135.5267 kJ/mol
* Info * Minimized energy of conformer 3: 155.2521 kJ/mol
* Info * Minimized energy of conformer 4: 178.1366 kJ/mol
* Info * Minimized energy of conformer 5: 145.9135 kJ/mol
* Info * Minimized energy of conformer 6: 182.1414 kJ/mol
* Info * Minimized energy of conformer 7: 186.1316 kJ/mol
* Info * Minimized energy of conformer 8: 160.9728 kJ/mol
* Info * Minimized energy of conformer 9: 152.4948 kJ/mol
* Info * Minimized energy of conformer 10: 169.1044 kJ/mol
* Info * Minimized energy of conformer 11: 143.8829 kJ/mol
* Info * Minimized energy of conformer 12: 169.7471 kJ/mol
* Info * Minimized energy of conformer 13: 144.0129 kJ/mol
* Info * Minimized energy of conformer 14: 145.2779 kJ/mol
* Info * Minimized energy of conformer 15: 127.2814 kJ/mol
* Info * Minimized energy of conformer 16: 157.8690 kJ/mol
* Info * Minimized energy of conformer 17: 170.8926 kJ/mol
* Info * Minimized energy of conformer 18: 155.9973 kJ/mol
* Info * Minimized energy of conformer 19: 153.8957 kJ/mol
* Info * Minimized energy of conformer 20: 149.6313 kJ/mol
* Info * Minimized energy of conformer 21: 177.8522 kJ/mol
* Info * Minimized energy of conformer 22: 129.9378 kJ/mol
* Info * Minimized energy of conformer 23: 141.4818 kJ/mol
* Info * Minimized energy of conformer 24: 170.8348 kJ/mol
* Info * Minimized energy of conformer 25: 164.7427 kJ/mol
* Info * Minimized energy of conformer 26: 152.5018 kJ/mol
* Info * Minimized energy of conformer 27: 158.5091 kJ/mol
* Info * Minimized energy of conformer 28: 159.8780 kJ/mol
* Info * Minimized energy of conformer 29: 141.3201 kJ/mol
* Info * Minimized energy of conformer 30: 159.5203 kJ/mol
* Info * Minimized energy of conformer 31: 140.4180 kJ/mol
* Info * Minimized energy of conformer 32: 164.5253 kJ/mol
* Info * Minimized energy of conformer 33: 168.4773 kJ/mol
* Info * Minimized energy of conformer 34: 155.1946 kJ/mol
* Info * Minimized energy of conformer 35: 155.6625 kJ/mol
* Info * Minimized energy of conformer 36: 155.5464 kJ/mol
* Info * Minimized energy of conformer 37: 151.7496 kJ/mol
* Info * Minimized energy of conformer 38: 147.2164 kJ/mol
* Info * Minimized energy of conformer 39: 157.7939 kJ/mol
* Info * Saving the 3 lowest energy conformations
* Info * Conformational sampling completed!
Show the structures
for mol, e in zip(conformer_dict['molecules'], conformer_dict['energies']):
mol.show()
print(f"{e:.2f} kJ/mol")127.28 kJ/mol
129.94 kJ/mol
135.53 kJ/mol