Molecular mechanics (MM) describes the potential energy of a molecular system using classical force fields, where atoms are treated as point charges connected by springs. The total energy is expressed as a sum of bonded terms (bonds, angles, dihedrals) and non-bonded terms (electrostatics, van der Waals), enabling the simulation of large systems at a fraction of the cost of quantum chemical methods. When combined with molecular dynamics (MD), MM force fields allow the time evolution of a system to be propagated, giving access to thermodynamic and structural properties via statistical sampling.
VeloxChem interfaces with the OpenMM library to perform MM/MD simulations Gracia Trivino et al. (2025). Force field topology files can be prepared directly within VeloxChem.
Force field parameters for arbitrary organic molecules are generated through the MMForceFieldGenerator class, which automatically assigns GAFF atom types and derives partial charges via a restrained electrostatic potential (RESP) fit that accounts for chemically equivalent atoms. This workflow provides a robust starting point for any molecule without requiring manual parameter assignment.
For cases where the default GAFF dihedral parameters do not accurately reproduce the torsional potential of key rotatable bonds, VeloxChem provides an automated reparameterization procedure. Dihedral barriers are refitted against QM relaxed potential energy scans, iteratively improving the MM description of conformational preferences until satisfactory agreement with the reference QM data is achieved.
Finally, once a satisfactory force field has been obtained, input files can be exported for use with GROMACS or OpenMM, enabling production-scale molecular dynamics simulations on a wide range of hardware.
Let us assume we have a molecule for which we wish to construct a molecular mechanics (MM) force field.
Force field generation¶
An initial force field is created based on calculated RESP charges and tabulated GAFF parameters.
import veloxchem as vlx
ff_gen = vlx.MMForceFieldGenerator()
ff_gen.create_topology(molecule)Force field reparameterization¶
Rotatable bonds have been identified and stored by the force field generator.
print(ff_gen.rotatable_bonds)[[14, 15], [21, 22], [28, 29], [29, 31], [31, 32]]
We will focus on the reparameterization of the barrier around the rotatable bond [21, 22], and make use of QM relaxed-scan reference data from file 16-21-22-27.xyz.
reparam_results = ff_gen.reparameterize_dihedrals(
rotatable_bond=(21, 22),
scan_file="../input_files/16-21-22-27.xyz",
visualize=True,
verbose=False,
)Initial GAFF Force-field
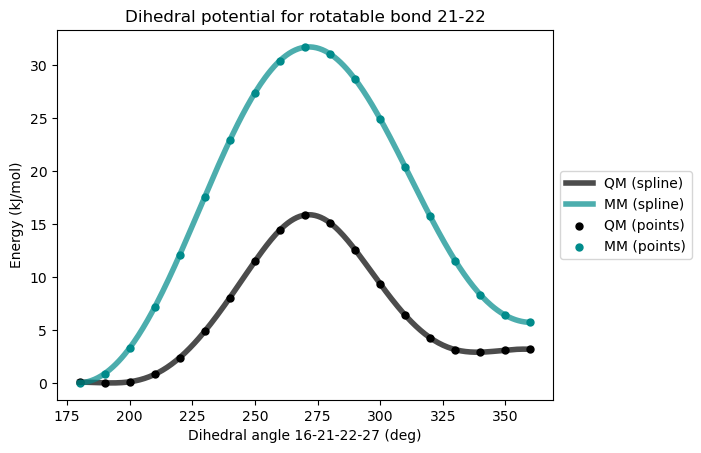
After first reparametrization
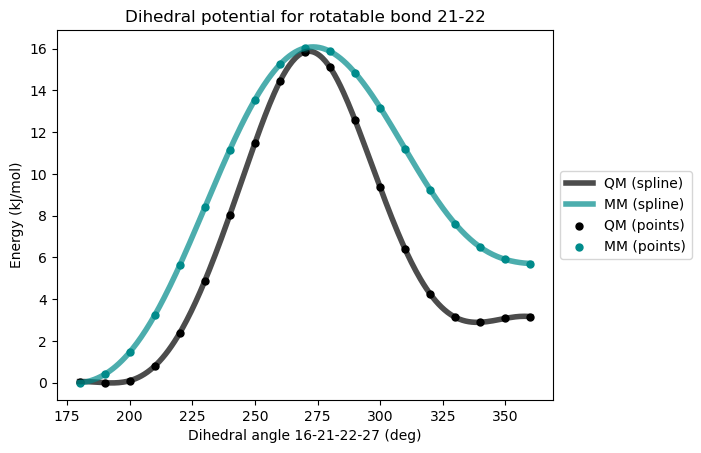
The rotational barrier is now much improved but the relative energies between the two conformer minima are still not well reproduced, leading to significant errors in the statistical dihedral distributions. As a remedy to this situation, an additional dihedral potential can be added.
ff_gen.add_dihedral((16, 21, 22, 27), barrier=0.0, phase=180.0, periodicity=1)We can now restart the reparametrization procedure with this added dihedral angle.
reparam_results = ff_gen.reparameterize_dihedrals(
rotatable_bond=(21, 22),
scan_file="../input_files/16-21-22-27.xyz",
visualize=True,
verbose=False,
initial_validation=False,
)After second reparametrization
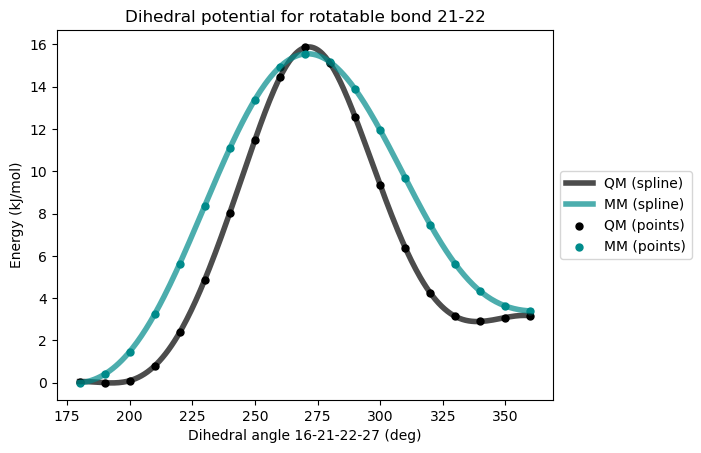
This time, both the barrier and the relative energy of the two minimun is well reproduced but the general shape of the potential energy surface can be improved to reach a better agreement with the QM potential. Another dihedral angle can be added for that purpose.
ff_gen.add_dihedral((16, 21, 22, 27), barrier=0.0, phase=0.0, periodicity=4)reparam_results = ff_gen.reparameterize_dihedrals(
rotatable_bond=(21, 22),
scan_file="../input_files/16-21-22-27.xyz",
visualize=True,
verbose=False,
initial_validation=False,
)After third reparametrization
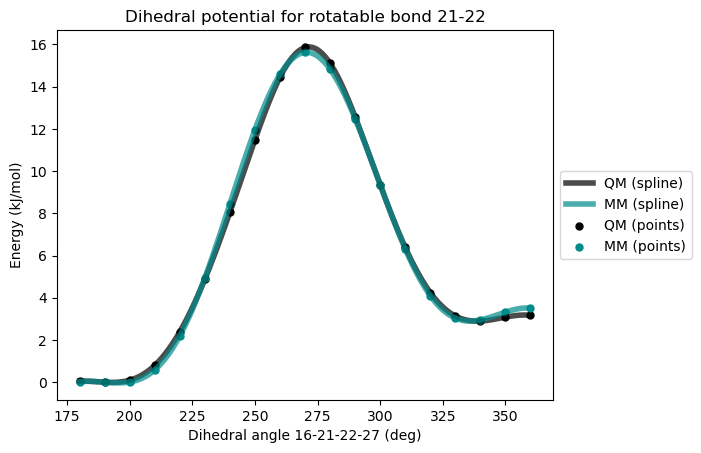
This time, the agreement betwwn the QM and MM potential is excellent. When a satisfactory quality has been reached in the force field, the files required to run GROMACS or OpenMM molecular dynamics simulations can be generated.
ff_gen.write_gromacs_files("HS-276_final", "MOL")
ff_gen.write_openmm_files("HS-276_final", "MOL")
- de Gracia Trivino, J. A., Brumboiu, I. E., Carrasco-Busturia, D., Li, X., Li, C., Linares, M., Lindfeld, V., Rhee, Y. M., Rune, J., Van Hoorn, B., Norman, P., & Ahlquist, M. S. G. (2025). VeloxChem Quantum–Classical Interoperability for Modeling of Complex Molecular Systems. J. Phys. Chem. A, 129(32), 7575–7587. 10.1021/acs.jpca.5c03187