Running on a laptop/desktop#
In a Jupyter notebook#
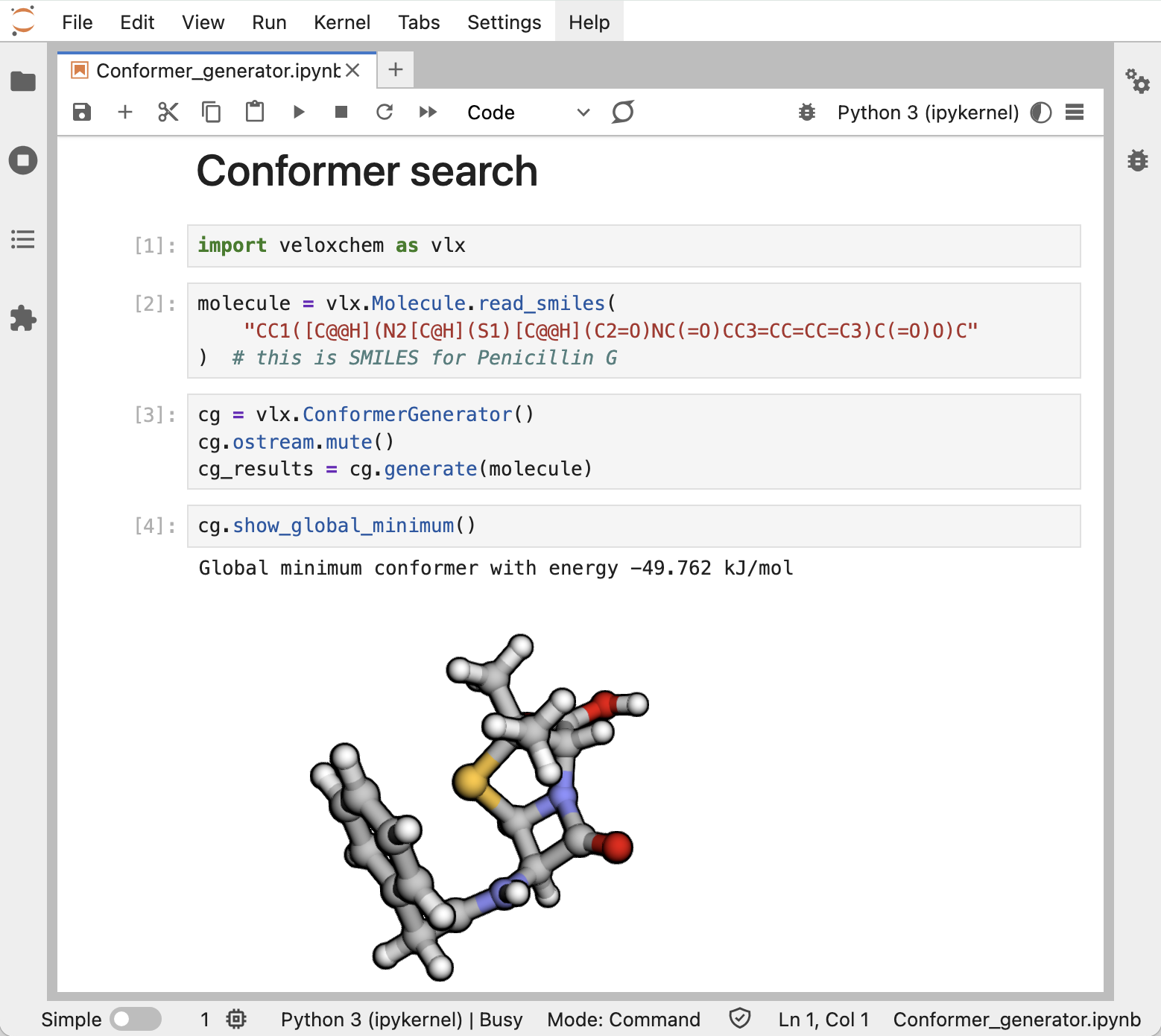
On a personal computer, we recommend importing the VeloxChem Python module and running calculations in Jupyter notebooks. This provides a very flexible framework for the creation of workflows, as amply illustrated in the eChem book [FDB+22].
Using an input file#
Calculations can also be run in the terminal window using input files in the form of Python scripts or text files.
Python script
Terminal command:
python myjob.py > myjob.out
The Python script input file named myjob.py above can e.g. take the form:
import veloxchem as vlx
xyz_string = """
3
water
O 0.0000000 0.0000000 -0.1653507
H 0.7493682 0.0000000 0.4424329
H -0.7493682 0.0000000 0.4424329
"""
molecule = vlx.Molecule.read_xyz_string(xyz_string)
basis = vlx.MolecularBasis.read(molecule, "def2-svp")
scf_drv = vlx.ScfRestrictedDriver()
scf_drv.xcfun = "b3lyp"
scf_drv.filename = "vlx_results_hdf5"
scf_results = scf_drv.compute(molecule, basis)
The results of the calculation are stored in an HDF5 file with a user specified name. This file can be directly read and analyzed with VIAMD.
Text file
Terminal command:
vlx myjob.inp [myjob.out]
An input file in text format consists of multiple groups marked with @group name and @end. The default unit for Cartesian coordinates of atoms is Angstrom. An example of the input file named myjob.inp above reads:
@jobs
task: scf
@end
@method settings
xcfun: b3lyp
basis: def2-svp
@end
@molecule
charge: 0
multiplicity: 1
xyz:
O 0.00000 0.00000 0.00000
H 0.00000 0.00000 1.79524
H 1.69319 0.00000 -0.59904
@end