VeloxChem enables the exploration of potential energy surfaces through efficient geometry optimizations and transition‑state searches, using the geomeTRIC module as a robust engine that provides stable structure updates for molecular systems.
Ground state optimization¶
VeloxChem evaluates analytic ground‑state gradients and employs quasi‑Newton optimization algorithms that approximate the local Hessian to achieve rapid convergence toward a minimum. For interactive visualisation of PES results, see this page.
Python script
import veloxchem as vlx
molecule = vlx.Molecule.read_name("bithiophene")
molecule.set_dihedral_in_degrees([3, 4, 6, 10], 160)
basis = vlx.MolecularBasis.read(molecule, "def2-svp")
scf_drv = vlx.ScfRestrictedDriver()
scf_drv.xcfun = "b3lyp"
scf_drv.dispersion = True
results = scf_drv.compute(molecule, basis)
opt_drv = vlx.OptimizationDriver(scf_drv)
opt_results = opt_drv.compute(molecule, basis, results)Reading bithiophene from PubChem...
Reference: S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang, E. E. Bolton, Nucleic Acids Res., 2025, 53, D1516-D1525.
Please double-check the compound since names may refer to more than one record.
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Kohn-Sham
Initial Guess Model : Superposition of Atomic Densities
Convergence Accelerator : Two Level Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Dispersion Correction : D4
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Using the D4 dispersion correction.
E. Caldeweyher, C. Bannwarth, S. Grimme, J. Chem. Phys., 2017, 147, 034112.
E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth, S. Grimme, J. Chem
Phys, 2019, 150, 154122.
* Info * Starting Reduced Basis SCF calculation...
* Info * ...done. SCF energy in reduced basis set: -1100.762094557229 a.u. Time: 0.76 sec.
Iter. | Kohn-Sham Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -1104.374454829720 0.0000000000 0.49183712 0.02685229 0.00000000
2 -1104.386796099115 -0.0123412694 0.39317655 0.02030594 0.44154018
3 -1104.406931505791 -0.0201354067 0.09963893 0.00611670 0.20970567
4 -1104.408096918369 -0.0011654126 0.03469937 0.00242192 0.05341051
5 -1104.408235402533 -0.0001384842 0.01352052 0.00090790 0.01827523
6 -1104.408261597186 -0.0000261947 0.00181768 0.00008929 0.00699465
7 -1104.408262029347 -0.0000004322 0.00095516 0.00004814 0.00158410
8 -1104.408262150381 -0.0000001210 0.00013680 0.00000672 0.00039059
9 -1104.408262152950 -0.0000000026 0.00006046 0.00000247 0.00008592
10 -1104.408262153417 -0.0000000005 0.00001843 0.00000068 0.00002932
11 -1104.408262153467 -0.0000000000 0.00000333 0.00000013 0.00001081
12 -1104.408262153468 -0.0000000000 0.00000104 0.00000006 0.00000332
13 -1104.408262153469 -0.0000000000 0.00000023 0.00000001 0.00000096
*** SCF converged in 13 iterations. Time: 9.85 sec.
Spin-Restricted Kohn-Sham:
--------------------------
Total Energy : -1104.4082621535 a.u.
Electronic Energy : -1740.1245052607 a.u.
Nuclear Repulsion Energy : 635.7526779259 a.u.
D4 Dispersion Correction : -0.0364348187 a.u.
------------------------------------
Gradient Norm : 0.0000002304 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
Optimization Driver Setup
===========================
Coordinate System : TRIC
Constraints : No
Max. Number of Steps : 300
Transition State : No
IRC : No
Hessian : never
* Info * Using geomeTRIC for geometry optimization.
L.-P. Wang and C.C. Song, J. Chem. Phys. 2016, 144, 214108
Optimization Step 0
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.456943000000 0.250181000000 -1.298614000000
C -2.434044000000 -1.090326000000 -0.936664000000
S -0.951126000000 -1.415293000000 -0.214158000000
C -0.328315000000 0.142071000000 -0.355598000000
C -1.282628000000 0.941060000000 -0.975504000000
C 1.000572000000 0.577542000000 0.101047000000
C 1.794312637373 -0.068473142343 1.042331877900
C 3.012842517545 0.580273710859 1.276978916616
C 3.184649532295 1.735274785992 0.525344016270
S 1.818868170862 1.948649318803 -0.431618302041
H -3.303202000000 0.716832000000 -1.784808000000
H -3.234074000000 -1.800918000000 -1.093817000000
H -1.140467000000 1.992011000000 -1.190436000000
H 1.507055037465 -0.981203180947 1.547615508776
H 3.751645975199 0.216001503034 1.978251653173
H 4.049459219367 2.384152819006 0.552836546709
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.011218687024 0.009927511100 0.002922620892
C 0.008257567484 -0.031101864386 0.010956859218
S -0.019726951770 0.049287119572 -0.020426441752
C -0.007016728346 -0.040578106410 0.009143343803
C -0.006275200927 -0.003166720656 -0.000522958380
C 0.011523833230 0.036278649425 -0.018176401977
C 0.005523965129 0.003876662267 0.002016929763
C -0.012601527897 -0.008608515349 -0.000154516026
C -0.002855123092 0.025966706236 -0.021764372305
S 0.010995006101 -0.040968399096 0.037895834840
H 0.005685477364 -0.002423437214 0.003038242004
H -0.000517608643 0.007509565952 -0.002593316859
H -0.002434002047 -0.006112828853 0.000002184053
H 0.003577428433 0.005020741457 -0.002295889520
H -0.005036906868 0.001807064870 -0.004331506418
H -0.000320198250 -0.006706821862 0.004273821266
*** Time spent in gradient calculation: 6.67 sec ***
* Info * Energy : -1104.4082621535 a.u.
* Info * Gradient : 2.873883e-02 a.u. (RMS)
* Info * 5.688244e-02 a.u. (Max)
* Info * Time : 18.72 sec
Optimization Step 1
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.459966487144 0.299857305859 -1.323552786456
C -2.335038061826 -1.016057200360 -0.904019298291
S -0.863289775214 -1.478110030071 -0.170084185818
C -0.335500937563 0.144220541917 -0.362586978852
C -1.262281452114 0.979124305556 -0.992668299992
C 0.977772523212 0.546922852139 0.103101253495
C 1.790728286908 -0.097672453475 1.041745347289
C 3.026929735949 0.536910141546 1.306635337503
C 3.070132121513 1.673689180023 0.518259780578
S 1.735676911202 1.965752999208 -0.506447332980
H -3.335029051992 0.733908926293 -1.811625591078
H -3.117773784896 -1.761658652946 -1.024320550023
H -1.083698146897 2.037244464107 -1.191470651854
H 1.481983927816 -1.017276098383 1.541779192482
H 3.796700532591 0.204878904172 2.006315811678
H 3.901646357062 2.374859889322 0.514763301462
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.016223051505 0.019282602213 -0.012551391529
C 0.039836348963 0.008774926184 0.015425035061
S 0.008401099476 -0.008540125380 0.004702805770
C -0.017210278690 -0.030454635025 0.001577594886
C -0.007407334436 0.005254821692 -0.004255129817
C 0.001241594658 0.019995168965 -0.012247994177
C 0.014305176729 0.010382368611 0.001338139848
C 0.017469996270 -0.017000484962 0.022144419552
C -0.036805159001 -0.010410777074 -0.013284598120
S -0.004372296762 0.000767604389 -0.004395753440
H -0.001158961113 0.002601450806 -0.001276718839
H 0.000764989982 0.003477693108 -0.000409745785
H -0.002183517621 0.000558483475 -0.001654540077
H 0.002692870762 0.000501938399 0.000598548146
H 0.001586692295 -0.001770655885 0.002120602581
H -0.000969192717 -0.003446010797 0.002172212371
*** Time spent in gradient calculation: 6.61 sec ***
* Info * Energy : -1104.4082286616 a.u.
* Info * Gradient : 2.219436e-02 a.u. (RMS)
* Info * 4.361039e-02 a.u. (Max)
* Info * Time : 17.44 sec
Optimization Step 2
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.449966557071 0.273218619043 -1.314727882407
C -2.404062256910 -1.049526861005 -0.928816311286
S -0.906884530958 -1.466629753004 -0.187807648984
C -0.326518300741 0.162270083738 -0.354760715117
C -1.257338352564 0.979527683689 -0.985843026127
C 0.988081359086 0.536728278947 0.125796172356
C 1.784606865841 -0.119322579551 1.059343667495
C 3.013045725396 0.564002784832 1.277521738729
C 3.135091308073 1.712429461454 0.524973443441
S 1.768248883124 1.968649696534 -0.484873888664
H -3.315169787856 0.723414918411 -1.807405548921
H -3.189732989953 -1.795571747311 -1.056545835378
H -1.086211744730 2.040600972638 -1.185567514478
H 1.482617635346 -1.039985035572 1.567447683332
H 3.781316652607 0.226097662394 1.976725549373
H 3.972822565719 2.411331456230 0.520510528060
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000900633451 0.003104424881 -0.001688501589
C 0.008541414716 -0.003014602579 0.005253960866
S 0.001191404385 -0.001103009221 -0.000259566155
C -0.002508913959 -0.011862210251 0.002954989534
C -0.000681005337 0.009295125707 -0.002549958345
C 0.000903946673 0.009419426232 -0.004756349250
C 0.002078853579 -0.007977941671 0.007401596922
C -0.002434876401 -0.002253942981 -0.000408736865
C -0.006918375068 0.003898574013 -0.006042490102
S 0.001557551191 0.000247280359 -0.000336839455
H -0.000981717599 0.000688390961 -0.000673271722
H -0.001780479267 0.000058645737 -0.000594364292
H 0.000275867585 0.002005192743 -0.000846606577
H -0.000446809142 -0.002484650206 0.001141521539
H 0.000518290851 -0.000522616298 0.000605328492
H 0.001555361791 0.000479199070 0.000794974398
*** Time spent in gradient calculation: 6.89 sec ***
* Info * Energy : -1104.4196803260 a.u.
* Info * Gradient : 6.824225e-03 a.u. (RMS)
* Info * 1.247953e-02 a.u. (Max)
* Info * Time : 16.39 sec
Optimization Step 3
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.488785000136 0.262222203634 -1.331624372123
C -2.463108514603 -1.059675227768 -0.971888958014
S -0.962812081883 -1.449320503802 -0.204295468814
C -0.326403247768 0.168753996677 -0.346132158215
C -1.287012448879 0.937267091481 -0.974312000541
C 0.993073471056 0.531670632917 0.143891750980
C 1.823789104846 -0.074640501306 1.065091213203
C 3.069040947250 0.574542899984 1.302277559380
C 3.180587522871 1.706517365205 0.534855820193
S 1.787356520822 1.962915588425 -0.457163552729
H -3.338973554941 0.733346550243 -1.826563847041
H -3.236622388862 -1.813480404322 -1.111792447169
H -1.125767067082 1.998403339036 -1.164230763297
H 1.524196010663 -0.986021761980 1.582388626348
H 3.833266097684 0.231370648487 2.001433623116
H 4.013372851187 2.407316094610 0.504843935253
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.003786315671 -0.000045796634 -0.001344750777
C -0.004807721464 -0.002192581935 -0.002334915284
S -0.004508099066 0.013377461288 -0.004676218128
C 0.011246742889 0.006437061620 0.001321819779
C -0.002242537103 -0.015168090053 0.003294334803
C -0.005237285087 -0.004699596304 -0.000091592151
C 0.005374200730 0.014557942728 -0.007342994950
C 0.007990075219 -0.003364390973 0.007387024354
C -0.002266900002 -0.002333932863 -0.000502745828
S -0.002182867348 -0.005574581480 0.003820027261
H -0.000971497795 -0.001143019964 0.000049907293
H -0.001214201851 0.000911467176 -0.000912745472
H 0.000409019189 -0.001827918901 0.000676235857
H 0.000372024132 0.001656212382 -0.000912250789
H 0.001279440752 0.000542379458 0.000553578033
H 0.000515489936 -0.001163839387 0.001016560772
*** Time spent in gradient calculation: 7.08 sec ***
* Info * Energy : -1104.4181678482 a.u.
* Info * Gradient : 8.801679e-03 a.u. (RMS)
* Info * 1.716786e-02 a.u. (Max)
* Info * Time : 16.80 sec
Optimization Step 4
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.471089703918 0.261754567120 -1.320925277694
C -2.431985481896 -1.060466287687 -0.954203994712
S -0.932053139222 -1.452561649478 -0.190411922877
C -0.328489646349 0.177041519961 -0.351859694728
C -1.279675879922 0.960157271679 -0.977854756108
C 0.988784172539 0.543813963249 0.132760176392
C 1.797251971157 -0.086280205006 1.057488712079
C 3.040458349466 0.567518660403 1.289335503547
C 3.174291136805 1.704539531519 0.531623776812
S 1.789361227428 1.975524176642 -0.465737827708
H -3.329196805930 0.719970387814 -1.814092661236
H -3.201584873465 -1.818924577201 -1.088535901530
H -1.122643547533 2.022093271887 -1.170947069267
H 1.488895236898 -0.998276202390 1.570009580133
H 3.799823838341 0.217636770252 1.989784997986
H 4.013482390610 2.398070654828 0.510343811928
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000622055523 0.000873595515 -0.000274033636
C -0.001237071908 -0.002368314707 -0.000679783829
S -0.002105448469 0.005068514362 -0.001366905434
C 0.004164041199 0.001104984728 0.000408948888
C -0.001193274738 -0.004397823652 0.001038347764
C -0.002372824690 -0.002051275269 -0.000412648661
C 0.001599350765 0.005788560038 -0.002989815055
C 0.002499508099 -0.001621803655 0.002956602199
C -0.000650139774 0.000134912907 -0.001329615551
S -0.000301633715 -0.002397425112 0.002479624248
H -0.000315849923 -0.000538371760 0.000165442622
H -0.000557049476 0.000496747712 -0.000508575973
H 0.000228321115 -0.000479542015 0.000250311171
H -0.000016128442 0.000552735552 -0.000431815356
H 0.000414045907 0.000424199193 0.000120715508
H 0.000445424693 -0.000612481604 0.000572125444
*** Time spent in gradient calculation: 7.28 sec ***
* Info * Energy : -1104.4203331174 a.u.
* Info * Gradient : 3.274451e-03 a.u. (RMS)
* Info * 6.708528e-03 a.u. (Max)
* Info * Time : 16.04 sec
Optimization Step 5
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.465060061627 0.263226025360 -1.318327372880
C -2.420019753420 -1.053693828236 -0.934958768700
S -0.911741171121 -1.454528372528 -0.190658510792
C -0.324625269019 0.182085125326 -0.357323090688
C -1.272364757619 0.966223033236 -0.987327062786
C 0.991934796921 0.549620033782 0.125873521357
C 1.787279006687 -0.100274853475 1.050808904311
C 3.025674757080 0.562801379890 1.281438612149
C 3.164554395284 1.708730241763 0.538980181989
S 1.792152097948 1.983248779897 -0.474950761626
H -3.326237409717 0.716520743143 -1.811432668851
H -3.189296667151 -1.815154947142 -1.053750521619
H -1.115259691461 2.027018477532 -1.187738956104
H 1.477197472987 -1.015512526966 1.557767950832
H 3.783648838251 0.208396289784 1.981333793223
H 4.000703930046 2.406235991168 0.527433467309
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000998004514 0.000817646813 -0.001050082932
C 0.000661926600 0.000599848704 0.000817141445
S 0.000345363584 0.000474585352 -0.000856849193
C 0.001262070207 -0.000079032431 0.001293147163
C -0.000705797308 -0.001907548864 0.000179243290
C 0.000655069008 0.000888518892 0.000348948674
C -0.000088022803 -0.001433737599 0.001115901514
C -0.000706326661 -0.000081365833 -0.000847994160
C -0.000824372905 0.000526024493 -0.000151299590
S 0.000811297119 0.000520402786 -0.000775756030
H -0.000265700333 -0.000103640185 -0.000163971597
H -0.000192728487 0.000214479702 -0.000007393881
H 0.000096776549 -0.000167652313 -0.000023383279
H -0.000119122241 -0.000350362940 0.000062941562
H -0.000004112318 -0.000007863562 -0.000066742177
H 0.000058097614 0.000072738415 0.000122515989
*** Time spent in gradient calculation: 6.93 sec ***
* Info * Energy : -1104.4206068567 a.u.
* Info * Gradient : 1.160120e-03 a.u. (RMS)
* Info * 2.041818e-03 a.u. (Max)
* Info * Time : 14.83 sec
Optimization Step 6
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.456865133405 0.260333874166 -1.313897795820
C -2.417561744742 -1.059182657881 -0.939027233229
S -0.913314547951 -1.454160629093 -0.183654016469
C -0.331326617780 0.185078923896 -0.357513111566
C -1.271774722536 0.978034171383 -0.986262026525
C 0.985789252116 0.548770159063 0.127433121190
C 1.785868696280 -0.095853898550 1.051321681641
C 3.027402824033 0.561741278973 1.282784801920
C 3.164717289779 1.703760435726 0.534951394635
S 1.785793547406 1.982988031046 -0.471260830254
H -3.317894262745 0.712264243350 -1.807941522276
H -3.184073602499 -1.822530401329 -1.063353336985
H -1.118105993650 2.039802924460 -1.185108811843
H 1.475979949212 -1.009137588758 1.561089197178
H 3.785377807076 0.205918509358 1.982052759180
H 4.001268892372 2.400396595578 0.516173685025
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003268586723 -0.001465481394 0.001940867917
C -0.001053878895 -0.001224499272 -0.000264430371
S -0.000311963013 -0.000891705222 0.000190159610
C -0.002504840773 -0.000342373510 -0.001197411666
C 0.000567361707 0.004242652096 -0.000917344841
C -0.001026915370 -0.000534015310 -0.000226482555
C 0.000558577592 0.000874125731 -0.000271707441
C 0.000831523399 -0.000300275406 0.000720214398
C -0.000654776700 -0.000777828367 0.000074260622
S -0.000357500703 0.000306754098 -0.000347296588
H 0.000333703738 -0.000053975775 0.000182915552
H 0.000031946126 -0.000338131783 0.000084737874
H 0.000198186811 0.000444204787 -0.000026889884
H 0.000024812923 0.000111073437 -0.000063835833
H 0.000132119888 0.000005034193 0.000101745687
H -0.000052089173 -0.000076479369 0.000019104329
*** Time spent in gradient calculation: 6.57 sec ***
* Info * Energy : -1104.4205613368 a.u.
* Info * Gradient : 1.819685e-03 a.u. (RMS)
* Info * 4.377616e-03 a.u. (Max)
* Info * Time : 15.11 sec
Optimization Step 7
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.463418246306 0.262311139832 -1.317330754248
C -2.418439174410 -1.056384899679 -0.940526371413
S -0.915215559793 -1.454446809947 -0.183692104763
C -0.329062128074 0.182851949838 -0.355745852146
C -1.273983619405 0.970595164027 -0.984821144359
C 0.988097548665 0.548411668092 0.128506557974
C 1.787290275338 -0.096289945269 1.052465042064
C 3.027890122554 0.563818698290 1.281993011316
C 3.168480612078 1.706282229055 0.535175489367
S 1.788171874751 1.982792295305 -0.469578798105
H -3.323620279253 0.715883436143 -1.811593164392
H -3.184392799961 -1.820110231059 -1.064924868468
H -1.119219581544 2.032130255320 -1.182949774690
H 1.478181432212 -1.009590996142 1.562695329069
H 3.785604291804 0.208097585347 1.981411453870
H 4.005403439411 2.402551791546 0.516756097714
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000033385100 0.000108485103 0.000118111544
C 0.000280041599 0.000122385640 -0.000092139102
S 0.000100187161 -0.000332774541 0.000282239725
C -0.000322712636 -0.000139025276 -0.000262011620
C -0.000149947126 0.000323527317 -0.000166702363
C -0.000320447458 -0.000605351266 0.000111498163
C -0.000009287587 0.000214106742 -0.000102653405
C -0.000202113943 0.000137280232 -0.000147844646
C 0.000474944472 0.000169332987 -0.000026985361
S 0.000077183398 -0.000180336067 0.000328804605
H -0.000031072175 0.000010922638 0.000013656322
H 0.000056138233 0.000010436150 -0.000008713803
H 0.000010675569 0.000012398821 0.000021413573
H -0.000064592665 0.000054464167 -0.000062435256
H 0.000000629671 0.000080787510 -0.000020084769
H 0.000051571697 -0.000006710240 0.000012146012
*** Time spent in gradient calculation: 7.02 sec ***
* Info * Energy : -1104.4206471383 a.u.
* Info * Gradient : 3.282084e-04 a.u. (RMS)
* Info * 6.939514e-04 a.u. (Max)
* Info * Time : 15.40 sec
Optimization Step 8
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.463175233097 0.261006991283 -1.317262741998
C -2.419989583608 -1.056657639284 -0.936712829274
S -0.913692414208 -1.451820324266 -0.184411757137
C -0.326368992694 0.184915565039 -0.356519610924
C -1.273199919239 0.969399054413 -0.986449148153
C 0.990733789291 0.552285278005 0.126273802885
C 1.787680782950 -0.096510250113 1.049898397681
C 3.028127080859 0.562552781694 1.282312885139
C 3.166554133327 1.707096047587 0.537841005744
S 1.790438780064 1.986804717004 -0.471218875955
H -3.323235091415 0.714246540450 -1.812022358789
H -3.186815140616 -1.820151789137 -1.057826095689
H -1.119166098080 2.030533629277 -1.187311451517
H 1.477499727047 -1.011062157572 1.557512358442
H 3.785540422547 0.205474844968 1.981436897625
H 4.003130175599 2.403811986311 0.522524926120
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000150488014 -0.000068409486 -0.000056671309
C -0.000426730179 -0.000277259985 0.000073510833
S -0.000140015314 0.000535597889 -0.000414356362
C 0.000567277116 0.000092333536 0.000393292433
C -0.000082958525 -0.000363622116 0.000139178935
C 0.000460230191 0.000558377803 0.000028725530
C -0.000074308573 -0.000228485828 0.000165829561
C 0.000249304825 -0.000301970780 0.000243583758
C -0.000772730155 -0.000174416018 -0.000133459374
S 0.000114917546 0.000481622630 -0.000544902518
H -0.000016865367 -0.000070649451 -0.000017181397
H -0.000086339842 0.000019741168 -0.000011087694
H 0.000053031777 -0.000053976349 0.000026015360
H 0.000022597587 -0.000079248000 0.000043320869
H 0.000011914414 -0.000060987515 0.000030801725
H -0.000042693348 -0.000026694362 0.000031742739
*** Time spent in gradient calculation: 6.82 sec ***
* Info * Energy : -1104.4206441948 a.u.
* Info * Gradient : 4.652465e-04 a.u. (RMS)
* Info * 8.033332e-04 a.u. (Max)
* Info * Time : 14.61 sec
Optimization Step 9
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.463048674057 0.261779474032 -1.317677412997
C -2.419482255475 -1.056112036050 -0.937902306613
S -0.915050680934 -1.453158411728 -0.182904520799
C -0.327875506318 0.183637337662 -0.355834629467
C -1.273116250902 0.969843088343 -0.986072772572
C 0.989256280605 0.549982371153 0.127785785910
C 1.788302206319 -0.096604328126 1.050928640787
C 3.028493380516 0.563673423782 1.281766017722
C 3.167176903644 1.707709207502 0.536872484788
S 1.788180339081 1.985074087335 -0.469295230201
H -3.322726131156 0.715343475235 -1.812799853086
H -3.186108571536 -1.819564666665 -1.060240596145
H -1.118344726656 2.031000738658 -1.186360003569
H 1.479699329550 -1.011000583035 1.559634844139
H 3.786490725680 0.207102000388 1.980480486683
H 4.003400014830 2.404832331919 0.519847324303
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000022874448 0.000031069517 -0.000024624891
C -0.000001607381 -0.000016545876 0.000038263577
S 0.000021025334 0.000002808008 -0.000056295660
C -0.000001774831 0.000007041798 0.000001462188
C -0.000047459828 -0.000007059892 0.000018189504
C -0.000040809745 -0.000056738962 0.000031535126
C 0.000012967750 0.000033579045 0.000028517323
C 0.000003117239 -0.000022720810 -0.000006394209
C -0.000026651395 0.000020580080 -0.000008943115
S 0.000044674245 -0.000002844462 -0.000031898269
H -0.000005492609 -0.000010600325 -0.000005485455
H -0.000009799658 0.000007219179 0.000000559983
H 0.000014901326 -0.000000451050 0.000006384128
H -0.000011488724 -0.000003928418 -0.000005704354
H 0.000004426480 0.000005683255 -0.000001848368
H 0.000006021046 -0.000006642969 0.000014676709
*** Time spent in gradient calculation: 6.86 sec ***
* Info * Energy : -1104.4206510875 a.u.
* Info * Gradient : 3.876847e-05 a.u. (RMS)
* Info * 7.667600e-05 a.u. (Max)
* Info * Time : 14.79 sec
Optimization Step 10
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.462857544842 0.261602402668 -1.317662243612
C -2.419227333855 -1.056112788211 -0.937377630176
S -0.915230978630 -1.452518423887 -0.181142177825
C -0.327786008472 0.184053437523 -0.355571369524
C -1.272935298547 0.969878310461 -0.986394962346
C 0.989365399629 0.550618673650 0.127835851706
C 1.788610843883 -0.096416306801 1.050472237054
C 3.028747680167 0.563926790393 1.281433108349
C 3.167234131281 1.708279477441 0.537000226725
S 1.787746099852 1.986301597573 -0.468376173204
H -3.322417115273 0.714954076330 -1.813176347832
H -3.185675220474 -1.819760353961 -1.059584999795
H -1.118234490234 2.030902635269 -1.187423910064
H 1.480291987782 -1.011132152278 1.558761790069
H 3.786900921885 0.206972347079 1.979782470699
H 4.003395979233 2.405466307712 0.519941251214
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000000311891 0.000013944164 0.000018013087
C 0.000017198562 -0.000013260914 -0.000023915873
S -0.000003901618 0.000001798014 0.000010700852
C 0.000004135045 -0.000014793307 -0.000024862975
C -0.000021874057 0.000008440441 0.000016051599
C 0.000018066832 -0.000021954527 0.000000993315
C -0.000024216966 -0.000001275033 0.000020882096
C -0.000013227193 0.000001522088 0.000004474480
C -0.000000613229 -0.000003189362 -0.000023022396
S 0.000016997572 0.000016262688 -0.000009686965
H -0.000002437487 -0.000001403587 0.000003637815
H 0.000000249737 0.000001747723 -0.000007170146
H 0.000003177073 -0.000000770030 0.000007684977
H -0.000007691926 0.000000252711 -0.000000663894
H -0.000003322087 0.000000240179 0.000002267521
H 0.000002209981 -0.000007002571 0.000003321692
*** Time spent in gradient calculation: 6.94 sec ***
* Info * Energy : -1104.4206513522 a.u.
* Info * Gradient : 2.070928e-05 a.u. (RMS)
* Info * 3.230497e-05 a.u. (Max)
* Info * Time : 13.17 sec
* Info * Geometry optimization completed.
Final Geometry (Angstroms)
============================
Atom Coordinate X Coordinate Y Coordinate Z
C -2.462857544842 0.261602402668 -1.317662243612
C -2.419227333855 -1.056112788211 -0.937377630176
S -0.915230978630 -1.452518423887 -0.181142177825
C -0.327786008472 0.184053437523 -0.355571369524
C -1.272935298547 0.969878310461 -0.986394962346
C 0.989365399629 0.550618673650 0.127835851706
C 1.788610843883 -0.096416306801 1.050472237054
C 3.028747680167 0.563926790393 1.281433108349
C 3.167234131281 1.708279477441 0.537000226725
S 1.787746099852 1.986301597573 -0.468376173204
H -3.322417115273 0.714954076330 -1.813176347832
H -3.185675220474 -1.819760353961 -1.059584999795
H -1.118234490234 2.030902635269 -1.187423910064
H 1.480291987782 -1.011132152278 1.558761790069
H 3.786900921885 0.206972347079 1.979782470699
H 4.003395979233 2.405466307712 0.519941251214
Summary of Geometry Optimization
==================================
Opt.Step Energy (a.u.) Energy Change (a.u.) Displacement (RMS, Max)
-------------------------------------------------------------------------------------
0 -1104.408262153467 0.000000000000 0.000e+00 0.000e+00
1 -1104.408228661594 0.000033491873 8.870e-02 1.533e-01
2 -1104.419680325986 -0.011451664392 4.873e-02 8.588e-02
3 -1104.418167848158 0.001512477828 5.341e-02 7.529e-02
4 -1104.420333117379 -0.002165269221 2.741e-02 4.239e-02
5 -1104.420606856683 -0.000273739304 1.872e-02 3.674e-02
6 -1104.420561336826 0.000045519857 8.774e-03 1.332e-02
7 -1104.420647138253 -0.000085801427 4.746e-03 8.060e-03
8 -1104.420644194820 0.000002943433 4.121e-03 7.359e-03
9 -1104.420651087513 -0.000006892694 2.119e-03 3.545e-03
10 -1104.420651352176 -0.000000264662 7.936e-04 1.753e-03
Statistical Deviation between
Optimized Geometry and Initial Geometry
=========================================
Internal Coord. RMS deviation Max. deviation
-----------------------------------------------------------
Bonds 0.030 Angstrom 0.064 Angstrom
Angles 3.278 degree 5.977 degree
Dihedrals 0.598 degree 1.203 degree
*** Time spent in Optimization Driver: 174.15 sec
* Info * Optimization results written to file: vlx_20260417_58cac48f.h5
opt_molecule = vlx.Molecule.read_xyz_string(opt_results["final_geometry"])
opt_molecule.show()opt_drv.plot_convergence(opt_results)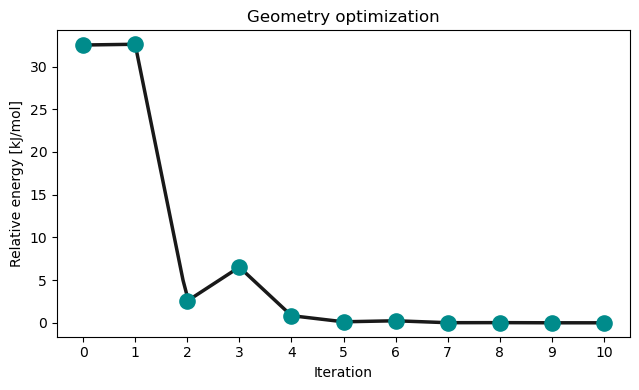
Text file
Please refer to the keyword list for a complete set of options. Dispersion can be activated in the @method settings section by using the keyword dispersion.
@jobs
task: optimize
@end
@method settings
xcfun: b3lyp
basis: def2-svp
dispersion: yes # use dft-d4 correction
@end
@molecule
charge: 0
multiplicity: 1
xyz:
...
@endConstrained optimization¶
Internal coordinates (distances, angles, dihedrals) can be constrained during the molecular structure optimization with use of either the set, freeze, or scan directives.
Set or freeze internal coordinate¶
The set directive will aim at converging an internal coordinate to a desired value while freeze will keep it at its initial value.
Python script
import veloxchem as vlx
molecule = vlx.Molecule.read_xyz_string("""4
Hydrogen peroxide
O -0.65564532 -0.06106286 -0.03621403
O 0.65564532 0.06106286 -0.03621403
H -0.97628735 0.65082652 0.57474201
H 0.97628735 -0.65082652 0.57474201
""")
basis = vlx.MolecularBasis.read(molecule, "def2-svp")
scf_drv = vlx.ScfRestrictedDriver()
scf_drv.xcfun = "b3lyp"
scf_results = scf_drv.compute(molecule, basis)
opt_drv = vlx.OptimizationDriver(scf_drv)
opt_drv.constraints = ["set dihedral 3 1 2 4 90.0", "freeze distance 1 2"]
opt_results = opt_drv.compute(molecule, basis, results)
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Kohn-Sham
Initial Guess Model : Superposition of Atomic Densities
Convergence Accelerator : Two Level Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Starting Reduced Basis SCF calculation...
* Info * ...done. SCF energy in reduced basis set: -150.566870205206 a.u. Time: 0.02 sec.
Iter. | Kohn-Sham Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -151.406804838447 0.0000000000 0.27077714 0.02660251 0.00000000
2 -151.411532718018 -0.0047278796 0.12427868 0.00997459 0.08009774
3 -151.412752069416 -0.0012193514 0.04230070 0.00328470 0.03847354
4 -151.412920062974 -0.0001679936 0.00152329 0.00013464 0.01144216
5 -151.412920268826 -0.0000002059 0.00024809 0.00002268 0.00052482
6 -151.412920273969 -0.0000000051 0.00002440 0.00000213 0.00007112
7 -151.412920274032 -0.0000000001 0.00000460 0.00000054 0.00000885
8 -151.412920274036 -0.0000000000 0.00000053 0.00000005 0.00000363
*** SCF converged in 8 iterations. Time: 0.14 sec.
Spin-Restricted Kohn-Sham:
--------------------------
Total Energy : -151.4129202740 a.u.
Electronic Energy : -190.4973689348 a.u.
Nuclear Repulsion Energy : 39.0844486607 a.u.
------------------------------------
Gradient Norm : 0.0000005312 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
Optimization Driver Setup
===========================
Coordinate System : TRIC
Constraints : Yes
Max. Number of Steps : 300
Transition State : No
IRC : No
Hessian : never
* Info * Using geomeTRIC for geometry optimization.
L.-P. Wang and C.C. Song, J. Chem. Phys. 2016, 144, 214108
Optimization Step 0
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.655645320000 -0.061062860000 -0.036214030000
O 0.655645320000 0.061062860000 -0.036214030000
H -0.976287350000 0.650826520000 0.574742010000
H 0.976287350000 -0.650826520000 0.574742010000
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.094374630664 -0.004996730482 -0.010571898169
O -0.094374630664 0.004996730482 -0.010571898169
H -0.003327137119 0.011492816581 0.010587064697
H 0.003327137119 -0.011492816581 0.010587064697
*** Time spent in gradient calculation: 0.11 sec ***
* Info * Energy : -151.4129202740 a.u.
* Info * Gradient : 6.818557e-02 a.u. (RMS)
* Info * 9.509628e-02 a.u. (Max)
* Info * Time : 0.33 sec
Optimization Step 1
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.657751160828 -0.031030129592 -0.062128633037
O 0.657751160828 0.031030129592 -0.062128633037
H -0.957953161752 0.617687114626 0.600014550564
H 0.957953161752 -0.617687114626 0.600014550564
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.088429554496 0.006395677486 -0.002076245647
O -0.088429554498 -0.006395677486 -0.002076245647
H -0.000409934106 -0.001257537135 0.002083624112
H 0.000409934106 0.001257537135 0.002083624112
*** Time spent in gradient calculation: 0.10 sec ***
* Info * Energy : -151.4130236485 a.u.
* Info * Gradient : 6.273393e-02 a.u. (RMS)
* Info * 8.868484e-02 a.u. (Max)
* Info * Time : 0.29 sec
Optimization Step 2
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.657704939711 -0.031994855305 -0.062334859026
O 0.657704939746 0.031994855306 -0.062334859021
H -0.955539766949 0.616538825990 0.599904242817
H 0.955539766974 -0.616538825989 0.599904242826
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.088403681080 0.006422331169 -0.001747710232
O -0.088403681080 -0.006422331169 -0.001747710234
H 0.000258341025 -0.001622416337 0.001754644528
H -0.000258341026 0.001622416337 0.001754644528
*** Time spent in gradient calculation: 0.09 sec ***
* Info * Energy : -151.4130258682 a.u.
* Info * Gradient : 6.271080e-02 a.u. (RMS)
* Info * 8.865389e-02 a.u. (Max)
* Info * Time : 0.28 sec
Optimization Step 3
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.657722872623 -0.031624062228 -0.062308908152
O 0.657722872663 0.031624062230 -0.062308908144
H -0.956417523289 0.616738798299 0.599650814138
H 0.956417523322 -0.616738798297 0.599650814148
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.088332921975 0.006634069232 -0.001664423754
O -0.088332921976 -0.006634069232 -0.001664423755
H 0.000084316812 -0.001691198938 0.001671148625
H -0.000084316813 0.001691198938 0.001671148625
*** Time spent in gradient calculation: 0.10 sec ***
* Info * Energy : -151.4130262190 a.u.
* Info * Gradient : 6.267035e-02 a.u. (RMS)
* Info * 8.859733e-02 a.u. (Max)
* Info * Time : 0.26 sec
* Info * Geometry optimization completed.
Final Geometry (Angstroms)
============================
Atom Coordinate X Coordinate Y Coordinate Z
O -0.657722872623 -0.031624062228 -0.062308908152
O 0.657722872663 0.031624062230 -0.062308908144
H -0.956417523289 0.616738798299 0.599650814138
H 0.956417523322 -0.616738798297 0.599650814148
Summary of Geometry Optimization
==================================
Opt.Step Energy (a.u.) Energy Change (a.u.) Displacement (RMS, Max)
-------------------------------------------------------------------------------------
0 -151.412920274036 0.000000000000 0.000e+00 0.000e+00
1 -151.413023648476 -0.000103374440 4.272e-02 4.556e-02
2 -151.413025868169 -0.000002219694 2.006e-03 2.699e-03
3 -151.413026219043 -0.000000350874 7.003e-04 8.899e-04
Statistical Deviation between
Optimized Geometry and Initial Geometry
=========================================
Internal Coord. RMS deviation Max. deviation
-----------------------------------------------------------
Bonds 0.015 Angstrom 0.018 Angstrom
Angles 1.131 degree 1.131 degree
Dihedrals 10.803 degree 10.803 degree
*** Time spent in Optimization Driver: 1.18 sec
* Info * Optimization results written to file: vlx_20260417_15cd2cda.h5
molecule_opt = vlx.Molecule.read_xyz_string(opt_results['final_geometry'])
molecule_opt.show(atom_indices=True)Text file
@jobs
task: optimize
@end
@method settings
xcfun: b3lyp
basis: def2-svp
@end
@optimize
constraints:
set dihedral 3 1 2 4 90.0
freeze distance 1 2
@end
@molecule
charge: 0
multiplicity: 1
xyz:
...
@endScan coordinate¶
The scan directive will perform a relaxed scan of an internal coordinate from an initial to a final value in a given number of steps.
scan distance 6 1 1.4 1.5 9
scan angle 6 1 2 100 110 9
scan dihedral 6 1 2 3 0 360 19Python script
import veloxchem as vlx
molecule = vlx.Molecule.read_xyz_string("""4
Hydrogen peroxide
O -0.65564532 -0.06106286 -0.03621403
O 0.65564532 0.06106286 -0.03621403
H -0.97628735 0.65082652 0.57474201
H 0.97628735 -0.65082652 0.57474201
""")
basis = vlx.MolecularBasis.read(molecule, "def2-svp")
scf_drv = vlx.ScfRestrictedDriver()
scf_drv.xcfun = "b3lyp"
scf_results = scf_drv.compute(molecule, basis)
opt_drv = vlx.OptimizationDriver(scf_drv)
opt_drv.constraints = ["scan dihedral 3 1 2 4 0 180 7"]
opt_results = opt_drv.compute(molecule, basis, scf_results)
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Kohn-Sham
Initial Guess Model : Superposition of Atomic Densities
Convergence Accelerator : Two Level Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Starting Reduced Basis SCF calculation...
* Info * ...done. SCF energy in reduced basis set: -150.566870205206 a.u. Time: 0.02 sec.
Iter. | Kohn-Sham Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -151.406804838447 0.0000000000 0.27077714 0.02660251 0.00000000
2 -151.411532718018 -0.0047278796 0.12427868 0.00997459 0.08009774
3 -151.412752069416 -0.0012193514 0.04230070 0.00328470 0.03847354
4 -151.412920062974 -0.0001679936 0.00152329 0.00013464 0.01144216
5 -151.412920268826 -0.0000002059 0.00024809 0.00002268 0.00052482
6 -151.412920273969 -0.0000000051 0.00002440 0.00000213 0.00007112
7 -151.412920274032 -0.0000000001 0.00000460 0.00000054 0.00000885
8 -151.412920274036 -0.0000000000 0.00000053 0.00000005 0.00000363
*** SCF converged in 8 iterations. Time: 0.13 sec.
Spin-Restricted Kohn-Sham:
--------------------------
Total Energy : -151.4129202740 a.u.
Electronic Energy : -190.4973689348 a.u.
Nuclear Repulsion Energy : 39.0844486607 a.u.
------------------------------------
Gradient Norm : 0.0000005312 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
Optimization Driver Setup
===========================
Coordinate System : TRIC
Constraints : Yes
Max. Number of Steps : 300
Transition State : No
IRC : No
Hessian : never
* Info * Using geomeTRIC for geometry optimization.
L.-P. Wang and C.C. Song, J. Chem. Phys. 2016, 144, 214108
Optimization Step 0
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.655645320000 -0.061062860000 -0.036214030000
O 0.655645320000 0.061062860000 -0.036214030000
H -0.976287350000 0.650826520000 0.574742010000
H 0.976287350000 -0.650826520000 0.574742010000
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.094374630664 -0.004996730482 -0.010571898169
O -0.094374630664 0.004996730482 -0.010571898169
H -0.003327137119 0.011492816581 0.010587064697
H 0.003327137119 -0.011492816581 0.010587064697
*** Time spent in gradient calculation: 0.11 sec ***
* Info * Energy : -151.4129202740 a.u.
* Info * Gradient : 6.818557e-02 a.u. (RMS)
* Info * 9.509628e-02 a.u. (Max)
* Info * Time : 0.41 sec
Optimization Step 1
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.607437155842 0.254202346373 -0.209988671444
O 0.607437155842 -0.254202346373 -0.209988671444
H -0.841050779934 0.351965765066 0.748516652179
H 0.841050779934 -0.351965765066 0.748516652179
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.102331159525 -0.042824200562 -0.023924693870
O -0.102331159524 0.042824200562 -0.023924693870
H 0.005605718971 -0.002345905961 0.023956402409
H -0.005605718971 0.002345905961 0.023956402409
*** Time spent in gradient calculation: 0.11 sec ***
* Info * Energy : -151.3960943561 a.u.
* Info * Gradient : 8.212431e-02 a.u. (RMS)
* Info * 1.134811e-01 a.u. (Max)
* Info * Time : 0.33 sec
Optimization Step 2
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.644940502122 0.276480296290 -0.187660008379
O 0.644940502114 -0.276480296290 -0.187660008382
H -0.949857245082 0.407195410565 0.725706265299
H 0.949857245078 -0.407195410565 0.725706265295
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.024415550116 -0.010466621167 0.001907820397
O -0.024415550117 0.010466621166 0.001907820397
H -0.006598061092 0.002828264751 -0.001899773205
H 0.006598061093 -0.002828264751 -0.001899773204
*** Time spent in gradient calculation: 0.11 sec ***
* Info * Energy : -151.4079142289 a.u.
* Info * Gradient : 1.955060e-02 a.u. (RMS)
* Info * 2.663286e-02 a.u. (Max)
* Info * Time : 0.31 sec
Optimization Step 3
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.662280528131 0.275644343086 -0.196933252370
O 0.662280528136 -0.275644343077 -0.196933252370
H -0.904560680755 0.376482508596 0.734572985182
H 0.904560680756 -0.376482508589 0.734572985182
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.006377283395 -0.002654312502 0.002661553934
O -0.006377283393 0.002654312503 0.002661553934
H 0.000086072316 -0.000036140017 -0.002660535145
H -0.000086072318 0.000036140017 -0.002660535147
*** Time spent in gradient calculation: 0.10 sec ***
* Info * Energy : -151.4097528263 a.u.
* Info * Gradient : 5.562650e-03 a.u. (RMS)
* Info * 7.402634e-03 a.u. (Max)
* Info * Time : 0.29 sec
Optimization Step 4
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.667957408889 0.276492107244 -0.199898808075
O 0.667957408886 -0.276492107271 -0.199898808065
H -0.900046871996 0.372562461625 0.737477911703
H 0.900046871998 -0.372562461643 0.737477911715
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.001540812565 -0.000638453416 -0.000055527911
O -0.001540812574 0.000638453421 -0.000055527914
H 0.000231766752 -0.000096213387 0.000061756693
H -0.000231766743 0.000096213383 0.000061756706
*** Time spent in gradient calculation: 0.12 sec ***
* Info * Energy : -151.4098666675 a.u.
* Info * Gradient : 1.194068e-03 a.u. (RMS)
* Info * 1.668775e-03 a.u. (Max)
* Info * Time : 0.30 sec
Optimization Step 5
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.669276154585 0.276993497972 -0.200112684428
O 0.669276154589 -0.276993498045 -0.200112684446
H -0.901360072160 0.373046099125 0.737389022675
H 0.901360072125 -0.373046099158 0.737389022653
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.000175452071 -0.000073220304 -0.000052906921
O -0.000175452089 0.000073220313 -0.000052906939
H 0.000066115869 -0.000027632213 0.000059359588
H -0.000066115852 0.000027632206 0.000059359620
*** Time spent in gradient calculation: 0.11 sec ***
* Info * Energy : -151.4098723347 a.u.
* Info * Gradient : 1.542760e-04 a.u. (RMS)
* Info * 1.973418e-04 a.u. (Max)
* Info * Time : 0.32 sec
Optimization Step 6
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.669416377797 0.277106235883 -0.200208303021
O 0.669416377861 -0.277106236016 -0.200208303063
H -0.901884397614 0.373336805897 0.737165341356
H 0.901884397545 -0.373336805932 0.737165341293
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O -0.000002638483 0.000000529835 -0.000002878612
O 0.000002638611 -0.000000529887 -0.000002878543
H 0.000004421486 -0.000002095445 0.000009320305
H -0.000004421613 0.000002095497 0.000009320118
*** Time spent in gradient calculation: 0.15 sec ***
* Info * Energy : -151.4098725056 a.u.
* Info * Gradient : 7.947840e-06 a.u. (RMS)
* Info * 1.052656e-05 a.u. (Max)
* Info * Time : 0.30 sec
Optimization Step 7
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.669416377797 0.277106235883 -0.200208303021
O 0.669416377861 -0.277106236016 -0.200208303063
H -0.901884397614 0.373336805897 0.737165341356
H 0.901884397545 -0.373336805932 0.737165341293
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O -0.000002638483 0.000000529835 -0.000002878612
O 0.000002638610 -0.000000529887 -0.000002878543
H 0.000004421486 -0.000002095445 0.000009320305
H -0.000004421613 0.000002095497 0.000009320118
*** Time spent in gradient calculation: 0.12 sec ***
* Info * Energy : -151.4098725056 a.u.
* Info * Gradient : 7.947840e-06 a.u. (RMS)
* Info * 1.052656e-05 a.u. (Max)
* Info * Time : 0.21 sec
Optimization Step 8
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.704828907728 0.167703929736 -0.184363617043
O 0.704828907474 -0.167703929752 -0.184363616833
H -0.893502305763 0.461973144397 0.721047953806
H 0.893502305986 -0.461973144565 0.721047954030
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O -0.000802368268 0.007555972431 0.000038373969
O 0.000802366111 -0.007555971175 0.000038372738
H -0.002334092369 -0.004604026963 -0.000030656786
H 0.002334094526 0.004604025706 -0.000030653583
*** Time spent in gradient calculation: 0.11 sec ***
* Info * Energy : -151.4123656260 a.u.
* Info * Gradient : 6.495536e-03 a.u. (RMS)
* Info * 7.598552e-03 a.u. (Max)
* Info * Time : 0.33 sec
Optimization Step 9
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.702821278059 0.165004123646 -0.187015292851
O 0.702821283230 -0.165004124715 -0.187015297129
H -0.876476654441 0.456344275264 0.723372892404
H 0.876476649228 -0.456344274366 0.723372886893
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.002014568197 0.005737190166 -0.001838936168
O -0.002014545835 -0.005737203670 -0.001838922200
H -0.000319724835 -0.004813035502 0.001847443156
H 0.000319702474 0.004813049007 0.001847407737
*** Time spent in gradient calculation: 0.12 sec ***
* Info * Energy : -151.4123988658 a.u.
* Info * Gradient : 5.789478e-03 a.u. (RMS)
* Info * 6.352601e-03 a.u. (Max)
* Info * Time : 0.30 sec
Optimization Step 10
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.703263011114 0.165906026386 -0.186264485508
O 0.703262981565 -0.165906020939 -0.186264453641
H -0.882757523756 0.458405825504 0.722386069285
H 0.882757553247 -0.458405831136 0.722386110391
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.001305038303 0.006301065655 -0.001286919947
O -0.001305069674 -0.006301047908 -0.001286931139
H -0.001096393875 -0.004695941278 0.001295404443
H 0.001096425256 0.004695923524 0.001295446240
*** Time spent in gradient calculation: 0.10 sec ***
* Info * Energy : -151.4124112395 a.u.
* Info * Gradient : 5.830724e-03 a.u. (RMS)
* Info * 6.562219e-03 a.u. (Max)
* Info * Time : 0.35 sec
Optimization Step 11
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.703154424785 0.165884576427 -0.186426945778
O 0.703154477239 -0.165884595601 -0.186426982940
H -0.882800290946 0.458411639777 0.722187860943
H 0.882800238000 -0.458411620494 0.722187828844
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.001419007727 0.006278654835 -0.001289736616
O -0.001418997832 -0.006278656989 -0.001289753466
H -0.001102644522 -0.004695251223 0.001298254649
H 0.001102634615 0.004695253382 0.001298258523
*** Time spent in gradient calculation: 0.12 sec ***
* Info * Energy : -151.4124112712 a.u.
* Info * Gradient : 5.832890e-03 a.u. (RMS)
* Info * 6.564949e-03 a.u. (Max)
* Info * Time : 0.32 sec
Optimization Step 12
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.703154424785 0.165884576427 -0.186426945778
O 0.703154477239 -0.165884595601 -0.186426982940
H -0.882800290946 0.458411639777 0.722187860943
H 0.882800238000 -0.458411620494 0.722187828844
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.001419007727 0.006278654835 -0.001289736616
O -0.001418997832 -0.006278656989 -0.001289753466
H -0.001102644522 -0.004695251223 0.001298254649
H 0.001102634615 0.004695253382 0.001298258523
*** Time spent in gradient calculation: 0.09 sec ***
* Info * Energy : -151.4124112712 a.u.
* Info * Gradient : 5.832890e-03 a.u. (RMS)
* Info * 6.564949e-03 a.u. (Max)
* Info * Time : 0.17 sec
Optimization Step 13
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.719340880948 0.067026392881 -0.139440411944
O 0.719340931970 -0.067026411574 -0.139440458573
H -0.916675368186 0.557784497295 0.675201331474
H 0.916675316672 -0.557784478492 0.675201300866
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O -0.003532393435 0.009665174197 -0.000857662081
O 0.003532416035 -0.009665184721 -0.000857668777
H -0.003162097114 -0.005401854938 0.000860641359
H 0.003162074505 0.005401865499 0.000860626315
*** Time spent in gradient calculation: 0.10 sec ***
* Info * Energy : -151.4176009679 a.u.
* Info * Gradient : 8.560045e-03 a.u. (RMS)
* Info * 1.032615e-02 a.u. (Max)
* Info * Time : 0.32 sec
Optimization Step 14
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.715325252075 0.057795945760 -0.143532055193
O 0.715325233798 -0.057795942078 -0.143532041993
H -0.885263639021 0.548060080124 0.679167266494
H 0.885263657199 -0.548060085240 0.679167262328
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.001884088616 0.004739427385 -0.003668955364
O -0.001884082730 -0.004739440870 -0.003668933727
H 0.001301765767 -0.004759710044 0.003678016077
H -0.001301771649 0.004759723532 0.003677991913
*** Time spent in gradient calculation: 0.09 sec ***
* Info * Energy : -151.4177255912 a.u.
* Info * Gradient : 6.218938e-03 a.u. (RMS)
* Info * 6.282770e-03 a.u. (Max)
* Info * Time : 0.27 sec
Optimization Step 15
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.716431440286 0.061150395847 -0.142275027571
O 0.716431420146 -0.061150393287 -0.142275007423
H -0.896907068397 0.551667729933 0.677663465465
H 0.896907088360 -0.551667734024 0.677663489646
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.000236829861 0.006435061555 -0.002820907773
O -0.000236836689 -0.006435058047 -0.002820906152
H -0.000417777219 -0.004900012512 0.002827750109
H 0.000417784053 0.004900008986 0.002827754778
*** Time spent in gradient calculation: 0.10 sec ***
* Info * Energy : -151.4177778995 a.u.
* Info * Gradient : 6.387661e-03 a.u. (RMS)
* Info * 7.030194e-03 a.u. (Max)
* Info * Time : 0.29 sec
Optimization Step 16
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.716239564599 0.061123963711 -0.142411499664
O 0.716239566561 -0.061123969666 -0.142411494262
H -0.896839835738 0.551639877873 0.677511514254
H 0.896839833347 -0.551639872711 0.677511536237
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.000441866597 0.006408545273 -0.002833851039
O -0.000441876612 -0.006408533820 -0.002833863243
H -0.000408566502 -0.004897458756 0.002840681002
H 0.000408576515 0.004897447294 0.002840700467
*** Time spent in gradient calculation: 0.12 sec ***
* Info * Energy : -151.4177780332 a.u.
* Info * Gradient : 6.384234e-03 a.u. (RMS)
* Info * 7.021069e-03 a.u. (Max)
* Info * Time : 0.32 sec
Optimization Step 17
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.716239564599 0.061123963711 -0.142411499664
O 0.716239566561 -0.061123969666 -0.142411494262
H -0.896839835738 0.551639877873 0.677511514254
H 0.896839833347 -0.551639872711 0.677511536237
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.000441866597 0.006408545273 -0.002833851039
O -0.000441876611 -0.006408533820 -0.002833863243
H -0.000408566502 -0.004897458756 0.002840681002
H 0.000408576515 0.004897447294 0.002840700467
*** Time spent in gradient calculation: 0.09 sec ***
* Info * Energy : -151.4177780332 a.u.
* Info * Gradient : 6.384234e-03 a.u. (RMS)
* Info * 7.021069e-03 a.u. (Max)
* Info * Time : 0.19 sec
Optimization Step 18
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.718672414704 -0.015660765747 -0.067182256394
O 0.718672415905 0.015660760660 -0.067182252632
H -0.954859941740 0.648815725219 0.602282270433
H 0.954859940111 -0.648815720925 0.602282295492
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O -0.001345960128 0.005726054102 -0.001322330962
O 0.001345958201 -0.005726048773 -0.001322336131
H -0.002971414578 -0.002303037408 0.001335753657
H 0.002971416510 0.002303032068 0.001335759009
*** Time spent in gradient calculation: 0.11 sec ***
* Info * Energy : -151.4217549881 a.u.
* Info * Gradient : 5.112013e-03 a.u. (RMS)
* Info * 6.028919e-03 a.u. (Max)
* Info * Time : 0.36 sec
Optimization Step 19
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.715951577635 -0.025103024610 -0.069684576059
O 0.715951583605 0.025103017116 -0.069684574054
H -0.931830083597 0.641679729160 0.604253183808
H 0.931830077007 -0.641679722028 0.604253200845
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.001894883433 0.001120200541 -0.002446915121
O -0.001894879420 -0.001120204806 -0.002446913333
H 0.001129058106 -0.001578694535 0.002459042551
H -0.001129062123 0.001578698804 0.002459038111
*** Time spent in gradient calculation: 0.10 sec ***
* Info * Energy : -151.4218695701 a.u.
* Info * Gradient : 3.213002e-03 a.u. (RMS)
* Info * 3.291326e-03 a.u. (Max)
* Info * Time : 0.29 sec
Optimization Step 20
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.717327010626 -0.022574712465 -0.069233497512
O 0.717327008658 0.022574707169 -0.069233487132
H -0.938262942343 0.643462725590 0.603423897482
H 0.938262943793 -0.643462720782 0.603423920997
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.000235614553 0.002562918545 -0.001975665465
O -0.000235639729 -0.002562889400 -0.001975679855
H 0.000019091665 -0.001958223099 0.001987963422
H -0.000019066488 0.001958193948 0.001987993782
*** Time spent in gradient calculation: 0.15 sec ***
* Info * Energy : -151.4218947077 a.u.
* Info * Gradient : 3.026077e-03 a.u. (RMS)
* Info * 3.244583e-03 a.u. (Max)
* Info * Time : 0.39 sec
Optimization Step 21
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.717512801935 -0.022767981247 -0.069517994506
O 0.717512859514 0.022767956906 -0.069518048934
H -0.938218629457 0.643265904908 0.603180755475
H 0.938218571286 -0.643265880829 0.603180724136
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.000032107813 0.002545543098 -0.001966434994
O -0.000032079212 -0.002545568566 -0.001966427173
H 0.000049336955 -0.001971209027 0.001978698326
H -0.000049365597 0.001971234564 0.001978671900
*** Time spent in gradient calculation: 0.10 sec ***
* Info * Energy : -151.4218948642 a.u.
* Info * Gradient : 3.012564e-03 a.u. (RMS)
* Info * 3.216797e-03 a.u. (Max)
* Info * Time : 0.30 sec
Optimization Step 22
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.717512801935 -0.022767981247 -0.069517994506
O 0.717512859514 0.022767956906 -0.069518048934
H -0.938218629457 0.643265904908 0.603180755475
H 0.938218571286 -0.643265880829 0.603180724136
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.000032107813 0.002545543098 -0.001966434994
O -0.000032079212 -0.002545568566 -0.001966427173
H 0.000049336955 -0.001971209027 0.001978698326
H -0.000049365597 0.001971234564 0.001978671900
*** Time spent in gradient calculation: 0.11 sec ***
* Info * Energy : -151.4218948642 a.u.
* Info * Gradient : 3.012564e-03 a.u. (RMS)
* Info * 3.216797e-03 a.u. (Max)
* Info * Time : 0.20 sec
Optimization Step 23
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.713982883318 -0.074642170398 0.028996472805
O 0.713982943243 0.074642152729 0.028996398824
H -0.998037430847 0.724036163757 0.504666299350
H 0.998037370330 -0.724036146350 0.504666267335
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.003016373920 0.002971918698 -0.000102246710
O -0.003016381477 -0.002971898601 -0.000102255194
H -0.003119548947 0.000366958122 0.000113961009
H 0.003119556486 -0.000366978143 0.000113971198
*** Time spent in gradient calculation: 0.09 sec ***
* Info * Energy : -151.4229608507 a.u.
* Info * Gradient : 3.729644e-03 a.u. (RMS)
* Info * 4.235713e-03 a.u. (Max)
* Info * Time : 0.30 sec
Optimization Step 24
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.713123612516 -0.083738939263 0.027663491257
O 0.713123671220 0.083738907598 0.027663418668
H -0.979750726521 0.718277429724 0.505500833873
H 0.979750668072 -0.718277401540 0.505500807124
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.003421883845 -0.001293257592 -0.000322435007
O -0.003421861574 0.001293229787 -0.000322424766
H 0.000925383150 0.000557012111 0.000332529442
H -0.000925405434 -0.000556984210 0.000332511773
*** Time spent in gradient calculation: 0.12 sec ***
* Info * Energy : -151.4230624481 a.u.
* Info * Gradient : 2.716875e-03 a.u. (RMS)
* Info * 3.672297e-03 a.u. (Max)
* Info * Time : 0.33 sec
Optimization Step 25
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.715472616722 -0.081506423937 0.027826835529
O 0.715472634097 0.081506395925 0.027826784575
H -0.984423624319 0.719653147409 0.504979633721
H 0.984423607646 -0.719653126188 0.504979615123
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.000807043695 0.000189076102 0.000030846122
O -0.000807030225 -0.000189099093 0.000030856690
H 0.000017078146 -0.000081678399 -0.000020849052
H -0.000017091629 0.000081701444 -0.000020863204
*** Time spent in gradient calculation: 0.10 sec ***
* Info * Energy : -151.4230933926 a.u.
* Info * Gradient : 5.896672e-04 a.u. (RMS)
* Info * 8.294702e-04 a.u. (Max)
* Info * Time : 0.31 sec
Optimization Step 26
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.716144079004 -0.081610155896 0.027536320408
O 0.716144062582 0.081610136590 0.027536285336
H -0.984573465418 0.719746620234 0.504771997247
H 0.984573483224 -0.719746610408 0.504771990991
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.000100842238 0.000097817621 0.000000801650
O -0.000100852509 -0.000097805486 0.000000798910
H -0.000012871537 -0.000051954467 0.000009219628
H 0.000012881805 0.000051942328 0.000009226935
*** Time spent in gradient calculation: 0.12 sec ***
* Info * Energy : -151.4230945689 a.u.
* Info * Gradient : 1.065069e-04 a.u. (RMS)
* Info * 1.404923e-04 a.u. (Max)
* Info * Time : 0.31 sec
Optimization Step 27
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.716249627201 -0.081718274248 0.027258017183
O 0.716249642033 0.081718248212 0.027257952501
H -0.984512189251 0.719712723183 0.504538789012
H 0.984512176010 -0.719712706591 0.504538774358
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O -0.000006581821 0.000025814032 -0.000021678209
O 0.000006563248 -0.000025782039 -0.000021689654
H 0.000000492897 -0.000018234314 0.000031718500
H -0.000000474333 0.000018202350 0.000031736949
*** Time spent in gradient calculation: 0.13 sec ***
* Info * Energy : -151.4230945899 a.u.
* Info * Gradient : 3.548042e-05 a.u. (RMS)
* Info * 3.658957e-05 a.u. (Max)
* Info * Time : 0.35 sec
Optimization Step 28
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.716249627201 -0.081718274248 0.027258017183
O 0.716249642033 0.081718248212 0.027257952501
H -0.984512189251 0.719712723183 0.504538789012
H 0.984512176010 -0.719712706591 0.504538774358
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O -0.000006581821 0.000025814032 -0.000021678209
O 0.000006563248 -0.000025782039 -0.000021689654
H 0.000000492897 -0.000018234314 0.000031718500
H -0.000000474333 0.000018202350 0.000031736949
*** Time spent in gradient calculation: 0.11 sec ***
* Info * Energy : -151.4230945899 a.u.
* Info * Gradient : 3.548042e-05 a.u. (RMS)
* Info * 3.658957e-05 a.u. (Max)
* Info * Time : 0.23 sec
Optimization Step 29
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.712301675600 -0.110985233026 0.142369061302
O 0.712301698314 0.110985217475 0.142368985718
H -1.027844740380 0.773010237281 0.389427751627
H 1.027844719257 -0.773010231174 0.389427737415
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.003825088879 0.002605111501 0.000642463209
O -0.003825066257 -0.002605141217 0.000642468003
H -0.002523109334 0.000506735774 -0.000634204867
H 0.002523086698 -0.000506706011 -0.000634213502
*** Time spent in gradient calculation: 0.13 sec ***
* Info * Energy : -151.4225352498 a.u.
* Info * Gradient : 3.798399e-03 a.u. (RMS)
* Info * 4.672330e-03 a.u. (Max)
* Info * Time : 0.35 sec
Optimization Step 30
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.712221793043 -0.117376284048 0.141861871193
O 0.712221777476 0.117376267910 0.141861814723
H -1.016606805814 0.769386793974 0.389569247766
H 1.016606823491 -0.769386789124 0.389569232621
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.003078203572 -0.000688202905 0.000597823483
O -0.003078195047 0.000688186064 0.000597826797
H 0.000369007845 0.000558032828 -0.000591113474
H -0.000369016373 -0.000558015985 -0.000591117884
*** Time spent in gradient calculation: 0.13 sec ***
* Info * Energy : -151.4226108513 a.u.
* Info * Gradient : 2.356194e-03 a.u. (RMS)
* Info * 3.210351e-03 a.u. (Max)
* Info * Time : 0.39 sec
Optimization Step 31
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.714557484189 -0.116556759292 0.141782897199
O 0.714557445567 0.116556748508 0.141782854181
H -1.018507905065 0.769985525062 0.389344140233
H 1.018507945971 -0.769985525922 0.389344127359
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.000499563493 0.000014428066 0.000720270573
O -0.000499598626 -0.000014373178 0.000720261607
H 0.000037929318 0.000017717298 -0.000713752918
H -0.000037894179 -0.000017772235 -0.000713737581
*** Time spent in gradient calculation: 0.12 sec ***
* Info * Energy : -151.4226283036 a.u.
* Info * Gradient : 7.999238e-04 a.u. (RMS)
* Info * 8.766882e-04 a.u. (Max)
* Info * Time : 0.31 sec
Optimization Step 32
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.715028773974 -0.116636144067 0.141585282057
O 0.715028835080 0.116636121515 0.141585180060
H -1.018702381073 0.770177493283 0.389206455783
H 1.018702321973 -0.770177480031 0.389206432693
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.000046234206 -0.000185891526 0.000676091877
O -0.000046203647 0.000185850126 0.000676098592
H -0.000027076811 0.000155846514 -0.000669510092
H 0.000027046229 -0.000155805000 -0.000669522373
*** Time spent in gradient calculation: 0.13 sec ***
* Info * Energy : -151.4226288555 a.u.
* Info * Gradient : 6.953614e-04 a.u. (RMS)
* Info * 7.027044e-04 a.u. (Max)
* Info * Time : 0.35 sec
Optimization Step 33
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.715028773974 -0.116636144067 0.141585282057
O 0.715028835080 0.116636121515 0.141585180060
H -1.018702381073 0.770177493283 0.389206455783
H 1.018702321973 -0.770177480031 0.389206432693
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.000046234206 -0.000185891526 0.000676091877
O -0.000046203647 0.000185850126 0.000676098592
H -0.000027076811 0.000155846514 -0.000669510092
H 0.000027046229 -0.000155805000 -0.000669522373
*** Time spent in gradient calculation: 0.10 sec ***
* Info * Energy : -151.4226288555 a.u.
* Info * Gradient : 6.953614e-04 a.u. (RMS)
* Info * 7.027044e-04 a.u. (Max)
* Info * Time : 0.22 sec
Optimization Step 34
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.713433934786 -0.126024398362 0.265395893534
O 0.713434010841 0.126024393419 0.265395769238
H -1.034408435743 0.788824086605 0.265395857956
H 1.034408361696 -0.788824090959 0.265395832079
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.001524787443 0.001257969796 -0.000000000645
O -0.001524754743 -0.001258015414 -0.000000000212
H -0.000929823050 0.000030572256 0.000000000496
H 0.000929790320 -0.000030526548 0.000000000364
*** Time spent in gradient calculation: 0.12 sec ***
* Info * Energy : -151.4222290376 a.u.
* Info * Gradient : 1.544821e-03 a.u. (RMS)
* Info * 1.976735e-03 a.u. (Max)
* Info * Time : 0.38 sec
Optimization Step 35
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.713419740996 -0.129268651396 0.265395886893
O 0.713419726315 0.129268640973 0.265395789446
H -1.029212415649 0.787030208561 0.265395849475
H 1.029212433585 -0.787030211232 0.265395826864
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.001071793028 -0.000524466681 -0.000000000033
O -0.001071786459 0.000524449491 0.000000000035
H 0.000486745920 0.000129060676 -0.000000000036
H -0.000486752487 -0.000129043489 0.000000000035
*** Time spent in gradient calculation: 0.11 sec ***
* Info * Energy : -151.4222379857 a.u.
* Info * Gradient : 9.157971e-04 a.u. (RMS)
* Info * 1.193233e-03 a.u. (Max)
* Info * Time : 0.31 sec
Optimization Step 36
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.714373399475 -0.128304215594 0.265395885518
O 0.714373368726 0.128304209704 0.265395790813
H -1.031184535459 0.787624532681 0.265395848621
H 1.031184569544 -0.787624540285 0.265395827710
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.000144840989 0.000086045298 -0.000000000016
O -0.000144856106 -0.000086023114 0.000000000016
H 0.000030101478 -0.000061455836 0.000000000002
H -0.000030086348 0.000061433612 -0.000000000002
*** Time spent in gradient calculation: 0.14 sec ***
* Info * Energy : -151.4222430261 a.u.
* Info * Gradient : 1.285770e-04 a.u. (RMS)
* Info * 1.684733e-04 a.u. (Max)
* Info * Time : 0.33 sec
Optimization Step 37
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
O -0.714509385810 -0.128373460959 0.265395891674
O 0.714509396832 0.128373456602 0.265395784658
H -1.031330952638 0.787622149535 0.265395849489
H 1.031330944442 -0.787622156929 0.265395826844
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
O 0.000010612870 0.000016693068 -0.000000000002
O -0.000010604921 -0.000016704136 0.000000000002
H 0.000001134933 -0.000007909720 0.000000000001
H -0.000001142883 0.000007920794 -0.000000000001
*** Time spent in gradient calculation: 0.09 sec ***
* Info * Energy : -151.4222430843 a.u.
* Info * Gradient : 1.508875e-05 a.u. (RMS)
* Info * 1.978617e-05 a.u. (Max)
* Info * Time : 0.25 sec
* Info * Geometry optimization completed.
Final Geometry (Angstroms)
============================
Atom Coordinate X Coordinate Y Coordinate Z
O -0.714509385810 -0.128373460959 0.265395891674
O 0.714509396832 0.128373456602 0.265395784658
H -1.031330952638 0.787622149535 0.265395849489
H 1.031330944442 -0.787622156929 0.265395826844
Summary of Geometry Scan
==========================
Scan Energy (a.u.) Relative Energy (a.u.) Relative Energy (kJ/mol)
----------------------------------------------------------------------------------
1 -151.409872505598 0.013222084293 34.7145775438
2 -151.412411271250 0.010683318641 28.0490492409
3 -151.417778033182 0.005316556709 13.9586177231
4 -151.421894864234 0.001199725657 3.1498792797
5 -151.423094589891 0.000000000000 0.0000000000
6 -151.422628855528 0.000465734363 1.2227854030
7 -151.422243084337 0.000851505554 2.2356275242
*** Time spent in Optimization Driver: 12.18 sec
* Info * Optimization results written to file: vlx_20260417_38a8cdfe.h5
opt_drv.plot_scan(opt_results)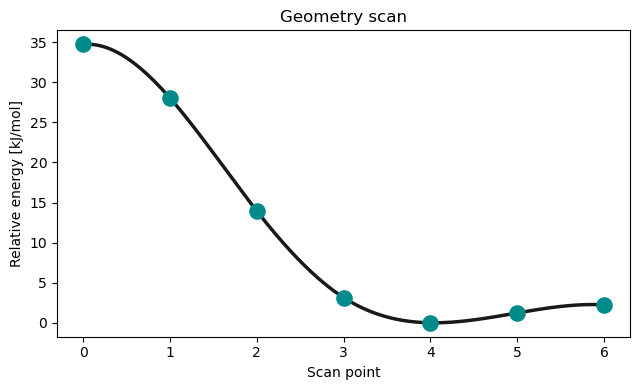
Text file
@jobs
task: optimize
@end
@method settings
xcfun: b3lyp
basis: def2-svp
@end
@optimize
constraints:
scan dihedral 3 1 2 4 180 0 7
@end
@molecule
charge: 0
multiplicity: 1
xyz:
...
@endTransition state optimization¶
Transition‑state optimization targets first‑order saddle points on the Born–Oppenheimer potential energy surface, characterized by vanishing gradients and a single imaginary Hessian eigenvalue. The resulting transition‑state geometry defines the critical point connecting reactant and product minima and provides the reference structure for calculating reaction pathways, activation energies, and vibrational signatures.
The optimization of a transition-state structure requires an initial guess of the structure that is close to the actual one.
Obtaining a start guess of the transition state structure¶
The transition-state guesser can be used to efficiently calculate an initial guess for a transition state structure. More information can be found in the eChem book
Python script
import veloxchem as vlx
reac1 = vlx.Molecule.read_smiles("C=CO")
reac2 = vlx.Molecule.read_smiles("C=O")
prod = vlx.Molecule.read_smiles("C(=O)CCO")
ts_guesser = vlx.TransitionStateGuesser()
ts_guesser.do_qm_scan = True
ts_guess_results = ts_guesser.find_transition_state([reac1, reac2], prod)* Info * Building forcefields. Disable mute_ff_build to see detailed output.
Reaction summary
1 breaking bonds:
ReaType ProType ID - ReaType ProType ID
oh o 3 - ho ho 7
2 forming bonds:
ReaType ProType ID - ReaType ProType ID
c2 c3 1 - c c3 8
ho ho 7 - o oh 9
* Info * System has charge 0.0 and multiplicity 1. Provide correct values if this is wrong.
* Info * Building MM systems for the transition state guess. Disable mute_ff_build to see detailed output.
Starting MM scan
MD steps: 1000
conf. snapshots: 1
MD temperature: 600 K
MD step size: 0.001 ps
folder name: ts_data_1780046220
saving MD traj: False
Lambda | E1 (kj/mol) | E2 (kj/mol) | V (kj/mol) | n_conf
------------------------------------------------------------
0.00 -14.26 1591.28 -14.26 1
0.05 -2.97 1291.24 61.74 1
0.10 17.75 1122.55 128.23 1
0.15 49.26 981.20 189.05 1
0.20 101.69 835.68 248.49 1
0.25 207.77 708.79 333.02 1
0.30 251.19 595.67 354.53 1
0.35 304.34 498.18 372.18 1
0.40 371.30 415.39 388.93 1
0.45 439.65 343.29 396.29 1
0.50 508.01 280.41 394.21 1
0.55 579.01 225.67 384.67 1
0.60 647.99 178.82 366.49 1
0.65 716.32 139.04 341.09 1
0.70 783.24 106.22 309.32 1
0.75 848.58 79.81 272.01 1
0.80 913.06 58.88 229.71 1
0.85 976.83 42.57 182.71 1
0.90 1040.64 30.20 131.24 1
0.95 1107.63 21.36 75.67 1
1.00 1198.20 15.39 15.39 1
* Info * Found highest MM E: 396.289 at Lambda: 0.45 and conformer index: 0.
* Info * Saving results to ts_results_1780046220.h5
* Info * Disable mute_scf to see detailed output.
Starting QM scan
QM parameters:
Basis: def2-svp
DFT xc fun: PBE0
Max conf.: 5
Lambda | Conf. i | Rel. E (kJ/mol) | MM V (kJ/mol)
------------------------------------------------------------
0.00 1 0.000 -14.261
0.05 1 12.378 61.738
0.10 1 22.923 128.233
0.15 1 35.562 189.051
0.20 1 55.396 248.490
0.25 1 107.000 333.022
0.30 1 93.949 354.535
0.35 1 82.195 372.183
0.40 1 73.848 388.934
0.45 1 60.140 396.289
0.50 1 42.245 394.213
0.55 1 22.696 384.672
0.60 1 1.676 366.489
0.65 1 -19.073 341.087
0.70 1 -38.782 309.323
0.75 1 -56.571 272.005
0.80 1 -72.071 229.713
0.85 1 -85.121 182.707
0.90 1 -95.636 131.242
0.95 1 -103.592 75.670
1.00 1 -109.309 15.393
* Info * Found highest QM E: -703034.304 at Lambda: 0.25 and conformer index: 0.
* Info * Saving results to ts_results_1780046220.h5
ts_guess=vlx.Molecule.read_xyz_string(ts_guess_results['max_qm_xyz'])
ts_guess.show()Optimizing the transition state structure¶
Python script
import veloxchem as vlx
ts_guess=vlx.Molecule.read_xyz_string(ts_guess_results['max_qm_xyz'])
basis = vlx.MolecularBasis.read(ts_guess, "def2-svp")
scf_drv = vlx.ScfRestrictedDriver()
scf_drv.xcfun = "B3LYP"
scf_results = scf_drv.compute(ts_guess, basis)
opt_drv = vlx.OptimizationDriver(scf_drv)
opt_drv.transition = True
opt_results = opt_drv.compute(ts_guess, basis, scf_results)
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Kohn-Sham
Initial Guess Model : Superposition of Atomic Densities
Convergence Accelerator : Two Level Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Starting Reduced Basis SCF calculation...
* Info * ...done. SCF energy in reduced basis set: -266.348854125709 a.u. Time: 0.15 sec.
Iter. | Kohn-Sham Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -268.050704726361 0.0000000000 0.50880603 0.03489599 0.00000000
2 -268.053119071087 -0.0024143447 0.48948825 0.02387564 0.27219669
3 -268.076765568835 -0.0236464977 0.14505013 0.00770965 0.15591440
4 -268.078687845989 -0.0019222772 0.03623327 0.00150643 0.05389749
5 -268.078830225286 -0.0001423793 0.01149562 0.00059418 0.01442717
6 -268.078844154819 -0.0000139295 0.00224509 0.00013188 0.00381992
7 -268.078844339774 -0.0000001850 0.00162813 0.00006963 0.00103956
8 -268.078844669056 -0.0000003293 0.00016549 0.00000780 0.00053539
9 -268.078844671321 -0.0000000023 0.00010306 0.00000472 0.00008995
10 -268.078844672905 -0.0000000016 0.00001572 0.00000081 0.00004397
11 -268.078844672943 -0.0000000000 0.00000275 0.00000011 0.00000738
12 -268.078844672943 -0.0000000000 0.00000042 0.00000002 0.00000136
*** SCF converged in 12 iterations. Time: 2.59 sec.
Spin-Restricted Kohn-Sham:
--------------------------
Total Energy : -268.0788446729 a.u.
Electronic Energy : -442.1837224271 a.u.
Nuclear Repulsion Energy : 174.1048777542 a.u.
------------------------------------
Gradient Norm : 0.0000004195 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
Optimization Driver Setup
===========================
Coordinate System : TRIC
Constraints : No
Max. Number of Steps : 300
Transition State : Yes
IRC : No
Hessian : first
* Info * Using geomeTRIC for geometry optimization.
L.-P. Wang and C.C. Song, J. Chem. Phys. 2016, 144, 214108
SCF Hessian Driver Setup
==========================
Hessian Type : Analytical
* Info * Computing analytical Hessian...
Reference: P. Deglmann, F. Furche, R. Ahlrichs, Chem. Phys. Lett. 2002, 362, 511-518.
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * CPHF/CPKS integral derivatives computed in 5.53 sec.
* Info * CPHF/CPKS right-hand side computed in 6.58 sec.
Coupled-Perturbed Kohn-Sham Solver Setup
------------------------------------------
Solver Type : Iterative Subspace Algorithm
Max. Number of Iterations : 150
Convergence Threshold : 1.0e-04
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * 30 trial vectors in reduced space
*** Iteration: 1 * Residuals (Max,Min): 4.99e-01 and 9.03e-02
* Info * 60 trial vectors in reduced space
*** Iteration: 2 * Residuals (Max,Min): 4.26e-02 and 1.12e-02
* Info * 90 trial vectors in reduced space
*** Iteration: 3 * Residuals (Max,Min): 3.40e-03 and 1.06e-03
* Info * 114 trial vectors in reduced space
*** Iteration: 4 * Residuals (Max,Min): 1.94e-04 and 6.65e-05
* Info * 140 trial vectors in reduced space
*** Iteration: 5 * Residuals (Max,Min): 9.29e-05 and 4.18e-06
*** Coupled-Perturbed Kohn-Sham converged in 5 iterations. Time: 37.19 sec
* Info * First order derivative contributions to the Hessian computed in 0.00 sec.
* Info * Second order derivative contributions to the Hessian computed in 30.70 sec.
*** Time spent in Hessian calculation: 67.89 sec ***
Optimization Step 0
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.958565365001 0.082810329601 -0.607450127181
C -1.266721765210 0.161564304416 0.737205612337
O -0.571318406706 -0.552635962856 1.581216123694
H -0.667255223825 -0.898103805146 -0.989570052974
H -1.527769932065 0.699085898972 -1.303754236807
H -2.265113517907 0.463899870329 1.071348490367
H 0.409312502108 -0.359313992243 1.284450564771
C 1.287844465926 0.704569952196 -0.410356738245
O 1.643992797005 0.079820002858 0.643629892952
H 1.725114973846 0.336050045752 -1.360438050652
H 1.213061740911 1.806920314360 -0.314034717792
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003644223188 -0.012370976715 0.006353145232
C 0.013103168591 0.042465939440 0.014732602702
O -0.005978154278 -0.017209502607 -0.005065349087
H -0.013746424870 -0.003584193246 -0.000545751933
H 0.002382018356 -0.000700171586 0.001894818688
H -0.003874496292 -0.022021450541 -0.008490788164
H -0.005294800584 0.012386918492 -0.007661939998
C -0.038883999557 0.010392247258 -0.038985250207
O 0.019275030339 -0.009054210363 0.022130012760
H 0.015080012400 0.000896815893 0.007807502067
H 0.014308442856 -0.001206535808 0.007864284596
*** Time spent in gradient calculation: 1.51 sec ***
* Info * Energy : -268.0788446729 a.u.
* Info * Gradient : 2.766799e-02 a.u. (RMS)
* Info * 5.603404e-02 a.u. (Max)
* Info * Time : 3.92 sec
Optimization Step 1
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.948776707582 0.085519823878 -0.612831394510
C -1.256716421050 0.159761476535 0.734679607276
O -0.566756241417 -0.548637660470 1.586001493508
H -0.661593466817 -0.895637269366 -0.997440417734
H -1.520685067205 0.703923438419 -1.305831846779
H -2.255832496756 0.465059586236 1.067840876388
H 0.421991505805 -0.360670238595 1.294805140380
C 1.282395876416 0.703295097499 -0.408332553255
O 1.633676334574 0.077727679633 0.646550026574
H 1.715698364823 0.333352289877 -1.360279869508
H 1.204481864769 1.806121088643 -0.315600779488
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.002514289272 -0.011859387421 0.004471842733
C 0.014322748162 0.041770950945 0.013987901813
O -0.005951113781 -0.016953849507 -0.003610390986
H -0.013593639523 -0.003443231106 -0.000443758252
H 0.002236817684 -0.000474095828 0.001542079643
H -0.004318946949 -0.021864573777 -0.007989300671
H -0.004484018213 0.011550471709 -0.006494832153
C -0.035596326255 0.010161227442 -0.036864628615
O 0.016991878938 -0.008649243056 0.020853288064
H 0.014450869715 0.000624147574 0.006998454003
H 0.013438523537 -0.000866444187 0.007581320646
*** Time spent in gradient calculation: 1.51 sec ***
* Info * Energy : -268.0800655041 a.u.
* Info * Gradient : 2.634373e-02 a.u. (RMS)
* Info * 5.224318e-02 a.u. (Max)
* Info * Time : 3.57 sec
Optimization Step 2
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.933697620223 0.089486742848 -0.619565376171
C -1.243228969356 0.157585191615 0.731314516456
O -0.560311906705 -0.543194708427 1.591104754046
H -0.651599474290 -0.891976393032 -1.007220685742
H -1.508696031558 0.710704408260 -1.308591871397
H -2.243145127195 0.467644770044 1.063336051276
H 0.439745058621 -0.362441642462 1.306847179263
C 1.274651780696 0.701290050104 -0.404981023796
O 1.619116173273 0.074593452260 0.650884417712
H 1.701207230420 0.329304934299 -1.359970950954
H 1.191364063352 1.804727863024 -0.317891214800
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.001108847992 -0.011372044798 0.002347094974
C 0.015889327328 0.040777195458 0.012991915082
O -0.005642159544 -0.016319010554 -0.002307630445
H -0.013240880410 -0.003255093092 -0.000348668462
H 0.001996881902 -0.000163631925 0.001138826673
H -0.004967764321 -0.021545399396 -0.007245818126
H -0.003548988018 0.010322851229 -0.004809981042
C -0.030517886175 0.009683029465 -0.033417417217
O 0.013414169709 -0.007979250716 0.018755471173
H 0.013436332141 0.000234321667 0.005826464302
H 0.012076394665 -0.000386916808 0.007100945328
*** Time spent in gradient calculation: 1.50 sec ***
* Info * Energy : -268.0816848425 a.u.
* Info * Gradient : 2.439588e-02 a.u. (RMS)
* Info * 4.627987e-02 a.u. (Max)
* Info * Time : 3.73 sec
Optimization Step 3
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.910254917646 0.095253887458 -0.627273858031
C -1.226227803280 0.155083385485 0.726865920742
O -0.551953125885 -0.535943968870 1.595380429192
H -0.633230079557 -0.886349200385 -1.018448221308
H -1.487857245600 0.720127296876 -1.312274408621
H -2.226518197182 0.473611614951 1.057740055437
H 0.463594959183 -0.364808600117 1.318339831266
C 1.263475468768 0.697875236785 -0.399173920368
O 1.600190597120 0.069993393516 0.657248689658
H 1.678332276493 0.323385382784 -1.359251739328
H 1.171021298133 1.801998559035 -0.321170490907
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000516008827 -0.011071597990 0.000314046499
C 0.017641449013 0.039295026954 0.011667996101
O -0.004762767210 -0.014970585588 -0.001694338704
H -0.012449737549 -0.002997194543 -0.000324667737
H 0.001600310256 0.000249598956 0.000763258903
H -0.005866874815 -0.020884420368 -0.006127230453
H -0.002687126836 0.008559071675 -0.002546838997
C -0.022797474243 0.008611239922 -0.027631793793
O 0.008090779821 -0.006753669605 0.015167857234
H 0.011776878837 -0.000279605870 0.004205260774
H 0.009966964827 0.000236890042 0.006239827774
*** Time spent in gradient calculation: 1.46 sec ***
* Info * Energy : -268.0838405840 a.u.
* Info * Gradient : 2.157393e-02 a.u. (RMS)
* Info * 4.462580e-02 a.u. (Max)
* Info * Time : 3.55 sec
Optimization Step 4
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.874887059503 0.102971354195 -0.634620727025
C -1.208362310187 0.151884870323 0.720505245999
O -0.543837321918 -0.526468359663 1.597443561293
H -0.598010373383 -0.877125720180 -1.029990488357
H -1.452093029717 0.732440569528 -1.317514936889
H -2.206433315670 0.488081277859 1.050543957621
H 0.491553148966 -0.369497575102 1.324502590018
C 1.248097180082 0.691888730884 -0.388644003624
O 1.581714280119 0.063774413820 0.666124814506
H 1.641128301347 0.315485248493 -1.357766845868
H 1.139165769916 1.796250139700 -0.325400271104
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.001948105905 -0.010978884951 -0.000984193828
C 0.018703072816 0.036676044166 0.009845454257
O -0.003378490757 -0.012410853511 -0.002250638850
H -0.010772517098 -0.002613834328 -0.000510927236
H 0.000977610458 0.000772013910 0.000589578533
H -0.006916953834 -0.019475542023 -0.004450472885
H -0.002164975768 0.006180728034 -0.000171023068
C -0.011966575630 0.006264339234 -0.018060395523
O 0.001636237837 -0.004431950370 0.009169979011
H 0.009016075669 -0.000830094407 0.002231002191
H 0.006816054386 0.000844551132 0.004630341386
*** Time spent in gradient calculation: 1.50 sec ***
* Info * Energy : -268.0869941480 a.u.
* Info * Gradient : 1.776917e-02 a.u. (RMS)
* Info * 4.233049e-02 a.u. (Max)
* Info * Time : 3.84 sec
Optimization Step 5
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.844025677319 0.106574216052 -0.638024665303
C -1.203793377203 0.145828856785 0.711513841485
O -0.545524646113 -0.516846565624 1.600014227417
H -0.546648077433 -0.865141195346 -1.038851452516
H -1.414647453484 0.739589438727 -1.324487188053
H -2.191431152826 0.516394391629 1.041841035692
H 0.502647481013 -0.383115614936 1.323569253641
C 1.243767541211 0.687013590643 -0.374001219864
O 1.583607492461 0.058442121087 0.672679769686
H 1.595456267968 0.309972202296 -1.358055499008
H 1.102245764080 1.788551354375 -0.326591395802
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000087748141 -0.009840278434 -0.001102203825
C 0.016445066367 0.031329897443 0.008624810615
O -0.005452775566 -0.008996473390 -0.002414670976
H -0.008767644924 -0.002281185917 -0.000980082298
H 0.000491163184 0.001322610018 0.000703633999
H -0.007491852980 -0.017004199831 -0.002721750139
H 0.000414312496 0.004391517579 -0.000446986283
C -0.003202244610 0.003463491076 -0.008619935761
O -0.001026673438 -0.002136819942 0.003717337120
H 0.005380047263 -0.001126478626 0.000852796830
H 0.003309143769 0.000872802598 0.002430040435
*** Time spent in gradient calculation: 1.51 sec ***
* Info * Energy : -268.0911429774 a.u.
* Info * Gradient : 1.402385e-02 a.u. (RMS)
* Info * 3.641964e-02 a.u. (Max)
* Info * Time : 3.46 sec
Optimization Step 6
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.832039327878 0.098560886681 -0.640360698810
C -1.208193697272 0.133947165060 0.700308498568
O -0.549136429527 -0.508317414111 1.605984374277
H -0.492096300368 -0.856131864751 -1.046341588683
H -1.394916524119 0.731265680214 -1.333739854072
H -2.173849299661 0.556810021362 1.031523566954
H 0.496579100486 -0.407942400520 1.325957016558
C 1.242723997662 0.686414376682 -0.361717993638
O 1.596012283899 0.055255917043 0.672328023916
H 1.558121643502 0.315435393491 -1.359359930959
H 1.076527822828 1.784199219903 -0.319674257608
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003091070555 -0.007317409867 -0.000058679090
C 0.011445225516 0.023990864746 0.006593289583
O -0.007148047449 -0.005569902968 -0.001354259353
H -0.007038356312 -0.001894683109 -0.001365219200
H 0.000477470637 0.001721245896 0.000791678773
H -0.007210025982 -0.013664368516 -0.001795839660
H 0.004170135158 0.003000299100 -0.001887508335
C -0.002297020761 0.001347402741 -0.004120990552
O -0.000505765413 -0.001113386611 0.001867781663
H 0.003150098787 -0.000805150172 0.000619063844
H 0.001871115022 0.000291994090 0.000754874067
*** Time spent in gradient calculation: 1.53 sec ***
* Info * Energy : -268.0949828894 a.u.
* Info * Gradient : 1.075318e-02 a.u. (RMS)
* Info * 2.738661e-02 a.u. (Max)
* Info * Time : 3.83 sec
Optimization Step 7
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.803963246414 0.093218768725 -0.645194647196
C -1.196015872337 0.122749778393 0.691583900896
O -0.541290362023 -0.501846260578 1.609032547414
H -0.444515146329 -0.853446207748 -1.051507125044
H -1.360705620955 0.726196577458 -1.343283899941
H -2.141556172888 0.591694396554 1.018517919735
H 0.506895804404 -0.423692267665 1.327989245990
C 1.204305763237 0.680009008851 -0.354112545313
O 1.591153295763 0.052162175294 0.674238844200
H 1.525341682767 0.325232736714 -1.355994087612
H 1.054029114121 1.780459022255 -0.312802290628
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000401120664 -0.005843393543 0.000429091071
C 0.009813788614 0.019042188597 0.002106384176
O -0.001678115792 -0.003320073319 -0.000866580610
H -0.004319635400 -0.000786650749 -0.001269930727
H 0.000263127721 0.001750828600 0.000766780060
H -0.006390420224 -0.010880837482 -0.001223601323
H 0.000249193955 0.000736841063 0.000020131326
C -0.003047288874 -0.001222884515 0.000226160998
O 0.000770662055 0.000977735487 -0.000487767330
H 0.002539715561 -0.000477738929 0.000453322666
H 0.002212992021 0.000007738340 -0.000105954711
*** Time spent in gradient calculation: 1.50 sec ***
* Info * Energy : -268.0970722369 a.u.
* Info * Gradient : 8.106066e-03 a.u. (RMS)
* Info * 2.152562e-02 a.u. (Max)
* Info * Time : 3.71 sec
Optimization Step 8
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.775728438061 0.084352363015 -0.643234923203
C -1.190195028260 0.111740746918 0.690141629954
O -0.537024202976 -0.501612673738 1.610281665311
H -0.409881316379 -0.862709032283 -1.042192356743
H -1.320521417283 0.717195382446 -1.350782910254
H -2.113730597889 0.626085506491 1.011314224340
H 0.518298392640 -0.427079969253 1.314570726031
C 1.190132224484 0.683401782441 -0.352246626196
O 1.575447569138 0.039793123901 0.673102886469
H 1.497179406628 0.337941788666 -1.360673812672
H 1.024602214126 1.781116675760 -0.300321901444
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000701292138 -0.005789357984 0.001489978609
C 0.006553090238 0.013969072063 0.001533859627
O 0.002656973650 -0.001400716417 -0.001554077443
H -0.002677564794 -0.000103198434 -0.001048154754
H 0.000622201397 0.001989765160 0.000809895011
H -0.005205568773 -0.008366444671 -0.000897989088
H -0.003047593957 -0.000302761414 0.000514278787
C 0.005045407970 0.001577409811 -0.000293868714
O -0.002832227687 -0.000379909102 0.000280053022
H -0.000175893187 -0.001197098167 -0.000228507421
H -0.000213744305 -0.000011281452 -0.000562142559
*** Time spent in gradient calculation: 1.45 sec ***
* Info * Energy : -268.0986155813 a.u.
* Info * Gradient : 6.375204e-03 a.u. (RMS)
* Info * 1.550583e-02 a.u. (Max)
* Info * Time : 3.60 sec
Optimization Step 9
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.797739775488 0.062151014126 -0.649552577805
C -1.201516923529 0.105609617563 0.681650888902
O -0.549284341391 -0.497587636440 1.616418669810
H -0.397037203467 -0.874733729590 -1.039067237720
H -1.344132887897 0.682443912190 -1.366150481553
H -2.095818473415 0.667874190185 1.001918585014
H 0.497827642593 -0.442511540340 1.323663396919
C 1.200496282580 0.688287971117 -0.347395884524
O 1.599280838920 0.027848106490 0.654463853566
H 1.499313129923 0.369680249644 -1.366907146526
H 1.020865341348 1.782538969937 -0.269604011414
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.002571579146 -0.002667569784 0.000375606317
C 0.003675671584 0.008296558130 0.003756358086
O -0.005001906097 -0.000579727681 0.000104582410
H -0.002831177686 -0.000542553061 -0.001021719738
H 0.000597593316 0.001621611619 0.000624137635
H -0.004234133730 -0.006188266646 -0.000875132067
H 0.003041714795 0.000575772811 -0.001896573835
C -0.002824988268 -0.000139386294 -0.002004618441
O 0.002301551710 -0.000218907076 0.001113055317
H 0.001257323354 -0.000318943362 0.000474364717
H 0.001459948269 0.000149777845 -0.000612060036
*** Time spent in gradient calculation: 1.50 sec ***
* Info * Energy : -268.0998240800 a.u.
* Info * Gradient : 4.686803e-03 a.u. (RMS)
* Info * 9.821083e-03 a.u. (Max)
* Info * Time : 3.78 sec
Optimization Step 10
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.777715985168 0.050670917069 -0.660154116039
C -1.183187592705 0.102232539987 0.671429512447
O -0.538200306148 -0.491317700625 1.614784094200
H -0.372369762577 -0.887834699944 -1.040645425506
H -1.323612135785 0.664283321172 -1.382618298930
H -2.055312788901 0.701961889535 0.986829470292
H 0.514005992047 -0.449206350166 1.322975781903
C 1.174374443142 0.688347159365 -0.339881269262
O 1.586866268147 0.016093992506 0.651715035055
H 1.475904184300 0.389794022576 -1.364768198068
H 0.995786692179 1.781564087361 -0.245723693581
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000045927001 -0.001388357003 -0.000107298060
C 0.004209836565 0.005686780713 0.000068202463
O 0.000262784607 0.000389939468 0.000550484961
H -0.001554868954 -0.000001822768 -0.000651006084
H 0.000423735770 0.001332184651 0.000482320512
H -0.003433533323 -0.004683439150 -0.000403736701
H -0.000155935412 -0.000741484946 0.000045923337
C -0.000919015209 -0.000669525468 0.000834186718
O -0.000120424482 0.000763050721 -0.000073220442
H 0.000281866636 -0.000743366107 -0.000020645518
H 0.001065553257 0.000040619484 -0.000686210371
*** Time spent in gradient calculation: 1.49 sec ***
* Info * Energy : -268.1005422753 a.u.
* Info * Gradient : 2.967317e-03 a.u. (RMS)
* Info * 7.075793e-03 a.u. (Max)
* Info * Time : 3.64 sec
Optimization Step 11
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.765905962372 0.031762011679 -0.660913751412
C -1.181817201488 0.099680057449 0.667970385611
O -0.536373502560 -0.487209460133 1.613167990918
H -0.360585758382 -0.913008498538 -1.026584670635
H -1.302493157680 0.638474242643 -1.396030155552
H -2.023321269185 0.744247746972 0.978039528045
H 0.519223691961 -0.445009554556 1.311017122996
C 1.164782183862 0.691411803718 -0.338078077885
O 1.585649676446 -0.006335013469 0.634999511978
H 1.466831501003 0.426100848901 -1.370983916127
H 0.966482145663 1.778108166547 -0.211902723264
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000677643872 -0.001760250984 0.001407776601
C 0.000297062148 0.002585485307 0.000166401647
O 0.002025019373 0.000831277101 -0.000409141060
H -0.001184878523 0.000040031319 -0.000584550955
H 0.000639726397 0.001303484746 0.000485398727
H -0.002038430341 -0.002821288790 -0.000145548989
H -0.002044310557 -0.000329495600 -0.000118707062
C 0.002249125232 0.000663973041 -0.000022521894
O -0.000466702468 0.000052798386 -0.000135916998
H -0.000267125014 -0.000669742825 0.000033824801
H 0.000129832421 0.000080144701 -0.000640979062
*** Time spent in gradient calculation: 1.44 sec ***
* Info * Energy : -268.1012157993 a.u.
* Info * Gradient : 2.011842e-03 a.u. (RMS)
* Info * 3.483684e-03 a.u. (Max)
* Info * Time : 3.56 sec
Optimization Step 12
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.771894792919 0.015871106003 -0.673431960602
C -1.169655902571 0.104456215184 0.658299502539
O -0.536091198807 -0.478063258549 1.616416430321
H -0.349831041344 -0.926303834176 -1.027247145073
H -1.317258985082 0.607239633511 -1.414208478520
H -1.990127507677 0.779530621030 0.961130055655
H 0.523042656167 -0.451362631746 1.322972118307
C 1.160637299452 0.694153560051 -0.333134312933
O 1.588330263284 -0.023786194815 0.620436786436
H 1.460113401060 0.456181484816 -1.373363589839
H 0.951059387000 1.774865003125 -0.178074216128
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000351934730 0.000530246965 -0.000698004895
C 0.002442974321 0.000894288044 0.000178985371
O -0.000741819692 0.000696716741 0.000997837080
H -0.000888937962 -0.000094181158 -0.000328878049
H 0.000228507069 0.000661768052 0.000136156258
H -0.001648153460 -0.001999710018 -0.000037471918
H 0.000418952917 -0.000575775447 0.000508369010
C -0.000322411502 0.000432931263 -0.000883383882
O -0.000253463594 -0.000155225246 0.000605520727
H 0.000028277182 -0.000520983010 -0.000052444194
H 0.000402315556 0.000103066070 -0.000390402403
*** Time spent in gradient calculation: 1.43 sec ***
* Info * Energy : -268.1016115158 a.u.
* Info * Gradient : 1.373390e-03 a.u. (RMS)
* Info * 2.607664e-03 a.u. (Max)
* Info * Time : 3.60 sec
Optimization Step 13
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.769691025738 -0.007995947469 -0.681188525729
C -1.172036805742 0.108024059940 0.646375395243
O -0.534349835977 -0.469736742493 1.604509432793
H -0.342323396876 -0.955429535924 -1.014640011890
H -1.303315413109 0.573792351343 -1.437661315551
H -1.958712422542 0.824620860646 0.942784255904
H 0.525091481731 -0.445758656223 1.299681326282
C 1.150549899000 0.696585499198 -0.323126533427
O 1.591630800020 -0.040316313581 0.609237088667
H 1.453745345862 0.493270202105 -1.369308339680
H 0.928862988986 1.770292446370 -0.137930871083
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000862825300 -0.000676820992 0.000992259222
C -0.002304983435 -0.000115726460 -0.000388664763
O 0.002107959872 0.000867601717 -0.000384963970
H -0.000599220637 -0.000079439311 -0.000291862445
H 0.000263994975 0.000512380950 0.000281470080
H -0.000299590758 -0.000619774883 0.000107771228
H -0.001204357429 0.000316492492 -0.001208324690
C -0.000527383279 -0.000538324346 0.001326216825
O 0.000780467397 0.000308245906 -0.000106012479
H 0.000241891258 -0.000198971540 0.000029491794
H 0.000692230833 0.000200699272 -0.000317638820
*** Time spent in gradient calculation: 1.41 sec ***
* Info * Energy : -268.1018791694 a.u.
* Info * Gradient : 1.385924e-03 a.u. (RMS)
* Info * 2.340385e-03 a.u. (Max)
* Info * Time : 3.33 sec
Optimization Step 14
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.761523045488 -0.019334344809 -0.688722800220
C -1.149718996053 0.117409039558 0.641352851405
O -0.533750106710 -0.458361676299 1.614782000348
H -0.329387928046 -0.968790403050 -1.010559727585
H -1.300912130301 0.550702531272 -1.450074932555
H -1.921636780798 0.856782468371 0.923385705022
H 0.535802362177 -0.450065843027 1.324274352931
C 1.141033571266 0.698564761701 -0.326139499193
O 1.590987522919 -0.064266627431 0.581981142336
H 1.439590441719 0.531998018990 -1.379223744137
H 0.895308115045 1.759545265305 -0.104094407721
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000395042835 0.001133959247 -0.000832856746
C 0.002217499882 -0.000930388432 0.000272835626
O -0.002444924828 0.000169904913 0.001002900280
H -0.000341569021 -0.000124349147 0.000017553942
H 0.000163387364 0.000144282167 -0.000180688776
H -0.000328831384 -0.000200495540 0.000023040699
H 0.000922037217 -0.000467866735 0.001816073720
C 0.000569616038 0.001096790657 -0.001592884021
O -0.000222629439 -0.000421846588 -0.000578936749
H -0.000262050778 -0.000168775127 0.000061239093
H -0.000647425220 -0.000250815970 0.000032867980
*** Time spent in gradient calculation: 1.42 sec ***
* Info * Energy : -268.1019366876 a.u.
* Info * Gradient : 1.505908e-03 a.u. (RMS)
* Info * 2.648081e-03 a.u. (Max)
* Info * Time : 3.50 sec
Optimization Step 15
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.769480952390 -0.030102769137 -0.694265413445
C -1.153455095947 0.117643746746 0.635329317022
O -0.525707751116 -0.460705079010 1.599090598274
H -0.330917804024 -0.979510668870 -1.007661731520
H -1.305741685040 0.534718199422 -1.461142342008
H -1.909843168607 0.871174905547 0.920772668043
H 0.540968117258 -0.449292699378 1.291454469880
C 1.135762232777 0.701884810822 -0.319797739744
O 1.587896664332 -0.059707678154 0.588478959161
H 1.441556500576 0.541118980945 -1.372139777871
H 0.893016695409 1.763016496445 -0.095031522830
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000509692193 -0.000247550853 0.000236932471
C -0.000923167713 0.000067760424 -0.000812478267
O 0.002835762643 0.000260544216 -0.000149870829
H 0.000027078609 -0.000008762745 0.000071487019
H 0.000060348885 -0.000099508599 0.000089342331
H 0.000269938446 0.000174963232 -0.000030062227
H -0.001376056411 -0.000112939102 -0.000682255782
C -0.000071225187 -0.000393236195 0.000906157219
O -0.000638602114 0.000228869672 0.000518739342
H 0.000175425899 0.000152258279 -0.000195131699
H 0.000162302886 -0.000039819144 0.000088994644
*** Time spent in gradient calculation: 1.41 sec ***
* Info * Energy : -268.1019962398 a.u.
* Info * Gradient : 1.142869e-03 a.u. (RMS)
* Info * 2.851648e-03 a.u. (Max)
* Info * Time : 3.51 sec
Optimization Step 16
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.768930584697 -0.018149249315 -0.695341544878
C -1.154550855939 0.113274188476 0.635505807787
O -0.531539430134 -0.477386852394 1.594952653544
H -0.333026968945 -0.964670345056 -1.020758726271
H -1.304715697523 0.557030629321 -1.455128664914
H -1.912693009388 0.862438444541 0.928324374506
H 0.537804115246 -0.458921980310 1.295242335933
C 1.139103889847 0.700730573224 -0.321497910160
O 1.589555636260 -0.066081155245 0.583067952458
H 1.439243728837 0.539977175380 -1.375103158138
H 0.899652903432 1.762154492050 -0.093955545670
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000033528187 0.000087670603 0.000032508109
C -0.000252507273 -0.000263372134 -0.000157470084
O 0.000342545429 -0.000039773864 -0.000021435617
H 0.000068586133 0.000030414031 0.000007091841
H -0.000001221225 -0.000015846128 -0.000002665553
H 0.000153428952 0.000154192903 0.000028907006
H -0.000413738102 0.000053930022 0.000209292874
C 0.000132502200 -0.000086681587 0.000060471531
O 0.000162028641 0.000041142110 -0.000134376341
H -0.000083913509 -0.000005695305 0.000002693927
H -0.000060041893 0.000023446788 0.000016914362
*** Time spent in gradient calculation: 1.45 sec ***
* Info * Energy : -268.1020246027 a.u.
* Info * Gradient : 2.422248e-04 a.u. (RMS)
* Info * 4.667881e-04 a.u. (Max)
* Info * Time : 3.35 sec
Optimization Step 17
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.771873657782 -0.016537873786 -0.699228477034
C -1.156958219551 0.115522718997 0.631662073959
O -0.533349783086 -0.473362113212 1.591810891285
H -0.335473670199 -0.962958442887 -1.024206632985
H -1.308416841957 0.557805326959 -1.459097915232
H -1.918495711927 0.861390557617 0.923906454476
H 0.535885663701 -0.455226197374 1.291868906470
C 1.139564250533 0.703723782947 -0.326042575215
O 1.586425811663 -0.060217611097 0.582462883064
H 1.440873820455 0.537777722868 -1.378576354841
H 0.900106712148 1.766010635062 -0.103183589011
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000049962098 0.000021468286 -0.000026213880
C -0.000022816737 -0.000106510590 -0.000111607457
O 0.000420668866 0.000063623057 0.000026716891
H -0.000034052351 -0.000005470969 0.000006700467
H 0.000015433077 0.000007512086 0.000002395541
H 0.000033147587 0.000009978464 0.000019075685
H -0.000207361757 -0.000090301719 0.000078503795
C 0.000259721235 0.000111729802 -0.000033349269
O -0.000332870786 -0.000006904026 0.000103710320
H -0.000038874137 -0.000017137418 -0.000029717342
H -0.000028483705 -0.000008688460 0.000005642692
*** Time spent in gradient calculation: 1.42 sec ***
* Info * Energy : -268.1020246140 a.u.
* Info * Gradient : 2.081011e-04 a.u. (RMS)
* Info * 4.262910e-04 a.u. (Max)
* Info * Time : 3.20 sec
Optimization Step 18
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.772409850878 -0.016293056819 -0.699474606782
C -1.157730726551 0.116228488849 0.631143465195
O -0.534396227222 -0.472525538314 1.591715893039
H -0.335243042923 -0.962436376895 -1.024214819994
H -1.308742472597 0.557862768490 -1.459634950370
H -1.919495863344 0.861972778432 0.923062234866
H 0.534714796913 -0.454845847322 1.292104277876
C 1.140217264374 0.703031266877 -0.325726628163
O 1.588025343567 -0.062267943289 0.580902292174
H 1.441286025803 0.539071726372 -1.378645527648
H 0.900599658664 1.764922762050 -0.101035772237
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000034579499 0.000070266364 0.000003682135
C -0.000068743458 -0.000127917932 0.000017944703
O -0.000038659358 0.000030170155 0.000014212108
H -0.000015517758 -0.000004110606 -0.000003029530
H 0.000000971913 0.000000580816 0.000005507332
H 0.000025186970 0.000015256045 0.000017309985
H -0.000017451199 0.000042269744 -0.000020306426
C -0.000076665191 -0.000038175742 0.000026913966
O 0.000131082202 -0.000010538055 -0.000015963023
H 0.000031598470 -0.000005734916 0.000007043174
H 0.000008089256 0.000007572060 -0.000011939178
*** Time spent in gradient calculation: 1.43 sec ***
* Info * Energy : -268.1020254880 a.u.
* Info * Gradient : 7.452088e-05 a.u. (RMS)
* Info * 1.463239e-04 a.u. (Max)
* Info * Time : 2.96 sec
Optimization Step 19
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.772206248395 -0.016434096868 -0.700635679983
C -1.157624142179 0.116691700218 0.629930069612
O -0.534771749693 -0.472057533376 1.590777149894
H -0.334883197498 -0.962653156131 -1.024922593811
H -1.308352079024 0.557531631886 -1.461079703193
H -1.919349306829 0.862668481717 0.921400518030
H 0.534705086383 -0.454816980252 1.291420902615
C 1.139768858836 0.703530479892 -0.326058007507
O 1.587155883988 -0.062426334892 0.580297008673
H 1.440795850994 0.540107465558 -1.379038105661
H 0.900306757095 1.765287583053 -0.100647468887
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000009445054 0.000003856521 -0.000008898947
C -0.000022344396 -0.000053834437 -0.000028404555
O 0.000051213725 0.000017768566 0.000015396269
H -0.000015109910 -0.000003054936 0.000002358611
H 0.000002509224 0.000006095440 0.000002059135
H 0.000010962990 0.000006036284 0.000016169571
H -0.000025409976 -0.000007109167 0.000035030059
C 0.000020292467 0.000032574774 0.000006256284
O -0.000022539012 -0.000011656043 -0.000004947457
H -0.000008407138 -0.000009270488 0.000000136230
H 0.000013691535 -0.000001704332 0.000006267198
*** Time spent in gradient calculation: 1.49 sec ***
* Info * Energy : -268.1020255484 a.u.
* Info * Gradient : 3.406155e-05 a.u. (RMS)
* Info * 6.484009e-05 a.u. (Max)
* Info * Time : 3.05 sec
* Info * Geometry optimization completed.
Final Geometry (Angstroms)
============================
Atom Coordinate X Coordinate Y Coordinate Z
C -0.772206248395 -0.016434096868 -0.700635679983
C -1.157624142179 0.116691700218 0.629930069612
O -0.534771749693 -0.472057533376 1.590777149894
H -0.334883197498 -0.962653156131 -1.024922593811
H -1.308352079024 0.557531631886 -1.461079703193
H -1.919349306829 0.862668481717 0.921400518030
H 0.534705086383 -0.454816980252 1.291420902615
C 1.139768858836 0.703530479892 -0.326058007507
O 1.587155883988 -0.062426334892 0.580297008673
H 1.440795850994 0.540107465558 -1.379038105661
H 0.900306757095 1.765287583053 -0.100647468887
Summary of Geometry Optimization
==================================
Opt.Step Energy (a.u.) Energy Change (a.u.) Displacement (RMS, Max)
-------------------------------------------------------------------------------------
0 -268.078844672943 0.000000000000 0.000e+00 0.000e+00
1 -268.080065504055 -0.001220831111 1.006e-02 1.489e-02
2 -268.081684842544 -0.001619338490 1.422e-02 1.912e-02
3 -268.083840584020 -0.002155741476 2.013e-02 2.943e-02
4 -268.086994148015 -0.003153563995 2.864e-02 4.712e-02
5 -268.091142977440 -0.004148829425 3.042e-02 5.611e-02
6 -268.094982889400 -0.003839911960 2.858e-02 4.811e-02
7 -268.097072236888 -0.002089347488 3.059e-02 4.756e-02
8 -268.098615581338 -0.001543344450 2.794e-02 4.503e-02
9 -268.099824079995 -0.001208498657 2.968e-02 4.931e-02
10 -268.100542275325 -0.000718195330 2.851e-02 5.482e-02
11 -268.101215799291 -0.000673523966 3.008e-02 5.516e-02
12 -268.101611515803 -0.000395716511 2.735e-02 5.102e-02
13 -268.101879169442 -0.000267653639 3.215e-02 5.869e-02
14 -268.101936687613 -0.000057518172 3.166e-02 5.254e-02
15 -268.101996239822 -0.000059552209 1.570e-02 2.983e-02
16 -268.102024602658 -0.000028362836 7.421e-03 1.115e-02
17 -268.102024614000 -0.000000011341 3.039e-03 5.045e-03
18 -268.102025488010 -0.000000874010 1.368e-03 2.996e-03
19 -268.102025548391 -0.000000060382 6.646e-04 9.723e-04
Statistical Deviation between
Optimized Geometry and Initial Geometry
=========================================
Internal Coord. RMS deviation Max. deviation
-----------------------------------------------------------
Bonds 0.024 Angstrom 0.068 Angstrom
Angles 2.328 degree 4.825 degree
Dihedrals 22.644 degree 38.517 degree
*** Time spent in Optimization Driver: 139.31 sec
* Info * Optimization results written to file: vlx_20260529_03a67682.h5
ts_opt = vlx.Molecule.read_xyz_string(opt_results['final_geometry'])
ts_opt.show(atom_indices=True)Text file
@jobs
task: optimize
@end
@method settings
xcfun: b3lyp
basis: def2-svp
dispersion: yes
@end
@optimize
transition: yes
@end
@molecule
charge: -1
multiplicity: 1
xyz:
...
@endts_opt = vlx.Molecule.read_xyz_string(opt_results['final_geometry'])
vib_drv = vlx.VibrationalAnalysis(scf_drv)
vib_drv.do_ir = False
vib_results = vib_drv.compute(ts_opt, basis)
Vibrational Analysis Driver
=============================
The following will be computed:
- Vibrational frequencies and normal modes
- Force constants
SCF Hessian Driver Setup
==========================
Hessian Type : Analytical
* Info * Computing analytical Hessian...
Reference: P. Deglmann, F. Furche, R. Ahlrichs, Chem. Phys. Lett. 2002, 362, 511-518.
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * CPHF/CPKS integral derivatives computed in 4.90 sec.
* Info * CPHF/CPKS right-hand side computed in 5.43 sec.
Coupled-Perturbed Kohn-Sham Solver Setup
------------------------------------------
Solver Type : Iterative Subspace Algorithm
Max. Number of Iterations : 150
Convergence Threshold : 1.0e-04
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * 30 trial vectors in reduced space
*** Iteration: 1 * Residuals (Max,Min): 4.90e-01 and 8.24e-02
* Info * 60 trial vectors in reduced space
*** Iteration: 2 * Residuals (Max,Min): 4.29e-02 and 7.86e-03
* Info * 90 trial vectors in reduced space
*** Iteration: 3 * Residuals (Max,Min): 3.18e-03 and 6.27e-04
* Info * 114 trial vectors in reduced space
*** Iteration: 4 * Residuals (Max,Min): 1.93e-04 and 4.54e-05
* Info * 138 trial vectors in reduced space
*** Iteration: 5 * Residuals (Max,Min): 9.85e-05 and 4.89e-06
* Info * Checkpoint written to file: vlx_20260417_a381fba0_orbrsp.h5
* Info * Time spent in writing checkpoint file: 0.01 sec
*** Coupled-Perturbed Kohn-Sham converged in 5 iterations. Time: 32.26 sec
* Info * First order derivative contributions to the Hessian computed in 0.00 sec.
* Info * Second order derivative contributions to the Hessian computed in 30.25 sec.
*** Time spent in Hessian calculation: 62.53 sec ***
Free Energy Analysis
======================
Note: Rotational symmetry is set to 1 regardless of true symmetry
1 Imaginary Frequencies (cm^-1): 809.136i
Note: Free energy does not include contribution from imaginary mode(s)
Free energy contributions calculated at @ 298.15 K:
Zero-point vibrational energy: 52.9154 kcal/mol
H (Trans + Rot + Vib = Tot): 1.4812 + 0.8887 + 1.4034 = 3.7733 kcal/mol
S (Trans + Rot + Vib = Tot): 38.8481 + 25.3752 + 7.3501 = 71.5734 cal/mol/K
TS (Trans + Rot + Vib = Tot): 11.5826 + 7.5656 + 2.1914 = 21.3396 kcal/mol
Ground State Electronic Energy : E0 = -268.10201889 au ( -168236.5569 kcal/mol)
Free Energy Correction (Harmonic) : ZPVE + [H-TS]_T,R,V = 0.05633243 au ( 35.3491 kcal/mol)
Gibbs Free Energy (Harmonic) : E0 + ZPVE + [H-TS]_T,R,V = -268.04568646 au ( -168201.2077 kcal/mol)
Vibrational Analysis
======================
* Info * The 5 lowest normal modes are printed below.
Vibrational Mode 1
----------------------------------------------------
Harmonic frequency: -809.14 cm**-1
Reduced mass: 2.0781 amu
Force constant: 0.8016 mdyne/A
Normal mode:
X Y Z
1 C 0.0849 -0.0221 -0.1470
2 C 0.0363 -0.0784 0.0162
3 O -0.0124 0.0374 -0.0390
4 H 0.0546 -0.0255 -0.0772
5 H -0.1070 -0.0054 0.1270
6 H -0.0111 0.0126 0.0178
7 H 0.3000 0.2407 -0.8267
8 C -0.1113 0.1006 0.1332
9 O -0.0153 -0.0472 0.0959
10 H -0.0182 0.0192 -0.0368
11 H 0.1023 -0.0877 -0.1362
Vibrational Mode 2
----------------------------------------------------
Harmonic frequency: 160.07 cm**-1
Reduced mass: 2.8545 amu
Force constant: 0.0431 mdyne/A
Normal mode:
X Y Z
1 C 0.0682 0.0620 0.0692
2 C 0.0441 -0.0130 0.0286
3 O -0.1522 -0.0541 -0.0059
4 H 0.2111 0.0834 0.0251
5 H -0.0517 0.1333 0.1817
6 H 0.1784 -0.0883 -0.0081
7 H -0.0337 0.0019 0.0095
8 C -0.1598 -0.0089 -0.1456
9 O 0.2159 0.0181 0.0625
10 H -0.2676 0.2858 -0.4669
11 H -0.4841 -0.3206 -0.0711
Vibrational Mode 3
----------------------------------------------------
Harmonic frequency: 302.41 cm**-1
Reduced mass: 2.4270 amu
Force constant: 0.1308 mdyne/A
Normal mode:
X Y Z
1 C 0.1737 0.0023 -0.0897
2 C -0.1483 -0.0531 0.1520
3 O 0.0825 -0.0029 0.0800
4 H 0.0650 -0.0213 0.0531
5 H 0.2353 0.0519 -0.2401
6 H -0.5939 -0.1405 0.5211
7 H 0.0130 0.0413 0.1177
8 C -0.0404 0.0230 -0.1305
9 O -0.0496 0.0278 -0.0314
10 H -0.0077 0.0355 -0.2525
11 H -0.0563 -0.0310 -0.1583
Vibrational Mode 4
----------------------------------------------------
Harmonic frequency: 388.98 cm**-1
Reduced mass: 3.0571 amu
Force constant: 0.2725 mdyne/A
Normal mode:
X Y Z
1 C 0.1203 -0.1789 -0.1650
2 C 0.1044 0.0403 -0.0450
3 O -0.0939 -0.0575 0.1737
4 H 0.0559 -0.1696 -0.4225
5 H 0.1387 -0.3533 0.0001
6 H 0.2652 0.1056 -0.2219
7 H -0.2714 -0.1553 0.3835
8 C -0.0653 0.0904 0.0709
9 O -0.0270 0.1099 -0.0640
10 H -0.0380 0.1081 -0.0008
11 H -0.1295 0.2081 0.1785
Vibrational Mode 5
----------------------------------------------------
Harmonic frequency: 504.71 cm**-1
Reduced mass: 4.3144 amu
Force constant: 0.6475 mdyne/A
Normal mode:
X Y Z
1 C -0.0600 -0.0693 0.0067
2 C 0.0721 -0.0682 -0.0814
3 O -0.0185 -0.0794 0.0321
4 H -0.3081 -0.1166 0.1805
5 H 0.1174 -0.1815 -0.1531
6 H 0.1768 0.0250 -0.2178
7 H 0.0665 0.0716 -0.1395
8 C 0.1678 0.0954 -0.2555
9 O -0.1458 0.1257 0.2870
10 H 0.2531 0.0518 -0.4449
11 H 0.1601 -0.0840 -0.3584
vib_drv.animate(vib_results, mode=1)Internal reaction coordinate¶
An intrinsic reaction coordinate (IRC) calculation can be performed to verify that a transition state structure connects the expected reactant and product structures.
Python script
import veloxchem as vlx
basis = vlx.MolecularBasis.read(ts_opt, "def2-svp")
scf_drv = vlx.ScfRestrictedDriver()
scf_drv.xcfun = "B3LYP"
scf_results = scf_drv.compute(ts_opt, basis)
opt_drv = vlx.OptimizationDriver(scf_drv)
opt_drv.irc = True
opt_results = opt_drv.compute(ts_opt, basis, scf_results)
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Kohn-Sham
Initial Guess Model : Superposition of Atomic Densities
Convergence Accelerator : Two Level Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Starting Reduced Basis SCF calculation...
* Info * ...done. SCF energy in reduced basis set: -266.370849678147 a.u. Time: 0.21 sec.
Iter. | Kohn-Sham Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -268.072462913249 0.0000000000 0.52427580 0.03559776 0.00000000
2 -268.077882570793 -0.0054196575 0.47540309 0.02903713 0.28531809
3 -268.100253731290 -0.0223711605 0.13451431 0.00641850 0.15238308
4 -268.101915608365 -0.0016618771 0.02830498 0.00126699 0.05026000
5 -268.102010243629 -0.0000946353 0.00842658 0.00033243 0.01311352
6 -268.102018314508 -0.0000080709 0.00236371 0.00012667 0.00317845
7 -268.102018694736 -0.0000003802 0.00131658 0.00006116 0.00095268
8 -268.102018891665 -0.0000001969 0.00012810 0.00000629 0.00042148
9 -268.102018893420 -0.0000000018 0.00004344 0.00000207 0.00005861
10 -268.102018893608 -0.0000000002 0.00002213 0.00000104 0.00002124
11 -268.102018893677 -0.0000000001 0.00000329 0.00000019 0.00000879
12 -268.102018893678 -0.0000000000 0.00000045 0.00000002 0.00000157
*** SCF converged in 12 iterations. Time: 2.45 sec.
Spin-Restricted Kohn-Sham:
--------------------------
Total Energy : -268.1020188937 a.u.
Electronic Energy : -447.8994148794 a.u.
Nuclear Repulsion Energy : 179.7973959857 a.u.
------------------------------------
Gradient Norm : 0.0000004497 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
Optimization Driver Setup
===========================
Coordinate System : TRIC
Constraints : No
Max. Number of Steps : 300
Transition State : No
IRC : Yes
Hessian : first
* Info * Using geomeTRIC for geometry optimization.
L.-P. Wang and C.C. Song, J. Chem. Phys. 2016, 144, 214108
SCF Hessian Driver Setup
==========================
Hessian Type : Analytical
* Info * Computing analytical Hessian...
Reference: P. Deglmann, F. Furche, R. Ahlrichs, Chem. Phys. Lett. 2002, 362, 511-518.
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * CPHF/CPKS integral derivatives computed in 4.70 sec.
* Info * CPHF/CPKS right-hand side computed in 6.36 sec.
Coupled-Perturbed Kohn-Sham Solver Setup
------------------------------------------
Solver Type : Iterative Subspace Algorithm
Max. Number of Iterations : 150
Convergence Threshold : 1.0e-04
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * 30 trial vectors in reduced space
*** Iteration: 1 * Residuals (Max,Min): 4.90e-01 and 8.24e-02
* Info * 60 trial vectors in reduced space
*** Iteration: 2 * Residuals (Max,Min): 4.29e-02 and 7.86e-03
* Info * 90 trial vectors in reduced space
*** Iteration: 3 * Residuals (Max,Min): 3.18e-03 and 6.27e-04
* Info * 114 trial vectors in reduced space
*** Iteration: 4 * Residuals (Max,Min): 1.93e-04 and 4.54e-05
* Info * 138 trial vectors in reduced space
*** Iteration: 5 * Residuals (Max,Min): 9.85e-05 and 4.89e-06
*** Coupled-Perturbed Kohn-Sham converged in 5 iterations. Time: 34.09 sec
* Info * First order derivative contributions to the Hessian computed in 0.00 sec.
* Info * Second order derivative contributions to the Hessian computed in 30.16 sec.
*** Time spent in Hessian calculation: 64.26 sec ***
Optimization Step 0
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.308752596435 0.949647816085 0.305888339275
C -0.547120658955 0.295808144813 1.269829093027
O -0.283575759200 -0.962693438609 1.203756811983
H -1.446178939858 2.030813582444 0.391608286651
H -2.107797571426 0.396341587025 -0.191212936627
H -0.008724184013 0.871470342657 2.044715751667
H -0.151800676874 -1.170579063602 0.120120909371
C -0.019855131876 0.405512208509 -1.229625153832
O 0.028125571743 -0.859902901128 -1.159330972062
H 0.865453221977 1.020227727634 -0.957976941271
H -0.668129181186 0.887738423186 -1.986739517579
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000005872557 -0.000000370503 0.000006541658
C -0.000023939270 -0.000036540706 0.000015927849
O -0.000026251016 -0.000019003474 0.000044966326
H -0.000007458418 -0.000003837435 0.000007043621
H 0.000019870043 -0.000002966259 -0.000013407191
H 0.000035083669 -0.000007525033 -0.000015338344
H 0.000036303210 -0.000013792907 0.000032412047
C 0.000052627184 0.000044239658 -0.000156594599
O -0.000049006903 -0.000023785513 0.000024370155
H -0.000014222865 0.000010707149 0.000040725182
H -0.000004195445 -0.000001017784 0.000008379848
*** Time spent in gradient calculation: 1.71 sec ***
* Info * Energy : -268.1020188937 a.u.
* Info * Gradient : 6.393942e-05 a.u. (RMS)
* Info * 1.710223e-04 a.u. (Max)
* Info * Time : 4.40 sec
Optimization Step 1
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.303402327919 0.948483258283 0.295368513748
C -0.544846181306 0.289127521069 1.271722871064
O -0.285396655523 -0.959705148786 1.202634436338
H -1.447371046927 2.028475485488 0.392369569144
H -2.123030013601 0.399327812280 -0.173320610041
H -0.008161499221 0.875404353288 2.043369692932
H -0.117720212471 -1.147818606359 0.040969039504
C -0.026593798992 0.413277161537 -1.221527156459
O 0.027144695412 -0.864195346226 -1.153537624889
H 0.865237624571 1.020532183176 -0.961830604906
H -0.656593385522 0.877887155954 -2.001101087524
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.003585020055 0.000848932971 0.008023392111
C -0.003005620783 0.005500551166 -0.001145548397
O 0.001331688977 -0.004710291262 -0.001237826636
H -0.000637363023 -0.000000969921 0.000525290781
H 0.000233131127 0.000340193280 -0.000168304018
H 0.000145361842 0.000034653400 -0.000315952252
H -0.001205675126 0.000208988623 0.007451744959
C 0.006941895871 -0.007294132436 -0.007176087177
O 0.000503272501 0.004896505383 -0.006884094917
H -0.000202353373 -0.000123726474 0.000120792756
H -0.000505126602 0.000258001332 0.000830817460
*** Time spent in gradient calculation: 1.69 sec ***
* Info * Energy : -268.1029049857 a.u.
* Info * Gradient : 6.235267e-03 a.u. (RMS)
* Info * 1.236489e-02 a.u. (Max)
* Info * Time : 4.30 sec
Optimization Step 2
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.298029347908 0.947158288461 0.284000066915
C -0.540769754101 0.281244203928 1.273363382807
O -0.286821888418 -0.955239969507 1.202895012947
H -1.440354086078 2.027564277242 0.384724876820
H -2.128807318445 0.397650538104 -0.166384629998
H -0.009864991890 0.876348855489 2.046451162034
H -0.094187518080 -1.137373638195 -0.064835253598
C -0.035666454222 0.423376216040 -1.211609696691
O 0.026293696281 -0.869333552169 -1.146453089691
H 0.865453758152 1.020569288450 -0.964708490691
H -0.646444982695 0.870917365270 -2.013405119062
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.009643640179 0.002516872929 0.020095737184
C -0.006551969554 0.013198676090 -0.003572740963
O 0.004869500067 -0.010097448199 -0.011378463606
H -0.000532393470 0.000167711596 0.000915836207
H 0.001191868605 -0.000127966481 -0.001540967331
H 0.000339465703 -0.000381197600 -0.000405200323
H -0.003929290419 0.001591215094 0.015016606125
C 0.015074504164 -0.017994143340 -0.016412625905
O 0.000962637462 0.009570308173 -0.005196364221
H 0.000082827759 0.000182684037 0.000702402141
H -0.001834463343 0.001339407365 0.001791779645
*** Time spent in gradient calculation: 1.71 sec ***
* Info * Energy : -268.1070709476 a.u.
* Info * Gradient : 1.409673e-02 a.u. (RMS)
* Info * 2.864270e-02 a.u. (Max)
* Info * Time : 4.31 sec
Optimization Step 3
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.291977554010 0.945507841974 0.271525387620
C -0.536721942266 0.273131614801 1.275481917784
O -0.289086749432 -0.950749097064 1.208036644815
H -1.436180546336 2.025923695908 0.378202865366
H -2.137814276408 0.399015410981 -0.154945563159
H -0.012198286545 0.879216145774 2.048925981718
H -0.065125251485 -1.143618576807 -0.168367425750
C -0.044910825709 0.434368821947 -1.201590259624
O 0.025797619786 -0.873835157468 -1.144228537830
H 0.864760786403 1.019686944418 -0.968675914998
H -0.633743021744 0.861942226314 -2.026448862170
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.015517033687 0.004505566345 0.030016179442
C -0.008508422892 0.015810164145 -0.005468671579
O 0.006772319911 -0.011090699408 -0.020437880305
H -0.000918828184 0.000342676106 0.001307507654
H 0.001902468259 -0.000100189592 -0.002254206979
H 0.000684597889 -0.000486699982 -0.000606190659
H -0.003191059668 0.007671896277 -0.003088111735
C 0.022845225338 -0.024144815627 -0.024383576884
O -0.001252402789 0.005974533996 0.021431075826
H -0.000386214547 -0.000117424730 0.000669370885
H -0.002407604068 0.001597463446 0.002864425489
*** Time spent in gradient calculation: 1.77 sec ***
* Info * Energy : -268.1119616292 a.u.
* Info * Gradient : 2.001152e-02 a.u. (RMS)
* Info * 4.122421e-02 a.u. (Max)
* Info * Time : 4.31 sec
Optimization Step 4
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.282349952024 0.941907113413 0.254664899103
C -0.532685589973 0.266101309603 1.277550233740
O -0.291746966400 -0.946968093279 1.217874468068
H -1.428040138893 2.022295054805 0.368623460062
H -2.147456058507 0.401077685224 -0.141356453565
H -0.016925850861 0.881118077552 2.052772036625
H -0.041958120562 -1.183905868014 -0.195627067160
C -0.058004767148 0.446378988681 -1.188173137958
O 0.026390988515 -0.875470428674 -1.150966804946
H 0.862111027812 1.019576418076 -0.969926668276
H -0.619940511782 0.855258290107 -2.039884114164
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.019920036783 0.007110005649 0.032081902431
C -0.005365805040 0.006339533365 -0.004521899194
O 0.004551061129 -0.002208802968 -0.021423658803
H -0.001349390971 0.000550875496 0.001561937184
H 0.001556157323 -0.000000990921 -0.002002021375
H 0.000866237521 -0.000171132339 -0.000686382769
H -0.002255990638 0.002195476880 -0.000455780958
C 0.025084969569 -0.018401815272 -0.027959856663
O -0.001080754966 0.004812122395 0.021276954401
H -0.000245858288 -0.001003620685 0.000033719123
H -0.001826790261 0.000753116764 0.002173822715
*** Time spent in gradient calculation: 1.71 sec ***
* Info * Energy : -268.1167230597 a.u.
* Info * Gradient : 1.979286e-02 a.u. (RMS)
* Info * 4.182865e-02 a.u. (Max)
* Info * Time : 4.21 sec
Optimization Step 5
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.270201584572 0.937140872143 0.234886222112
C -0.529193259245 0.261331240923 1.280431862855
O -0.294118748242 -0.944921704036 1.228444100080
H -1.417135414799 2.017680393480 0.355560280103
H -2.156112236176 0.403074275998 -0.127419163172
H -0.023394171670 0.883236174566 2.058400272813
H -0.022599643458 -1.211110140332 -0.203464689230
C -0.073119890179 0.457812294264 -1.171674277148
O 0.026971504440 -0.876988580472 -1.160534602365
H 0.856623217117 1.024520498726 -0.968424579864
H -0.607043966919 0.850337489663 -2.050836931049
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.023149478298 0.009653646792 0.032315299147
C -0.002317952804 -0.000956901827 -0.002966214058
O 0.002536378397 0.003722754243 -0.021404533087
H -0.001788996091 0.000769774646 0.001921635890
H 0.000654295108 -0.000080280312 -0.001690816228
H 0.001075675962 0.000122233004 -0.000851133191
H -0.001680451059 -0.003832107662 0.010791561428
C 0.025892184063 -0.013633284448 -0.027288702370
O -0.000494036013 0.005117351649 0.009553897061
H 0.000743634550 -0.000859021941 -0.000648600571
H -0.001455594477 -0.000037546156 0.000330351050
*** Time spent in gradient calculation: 1.72 sec ***
* Info * Energy : -268.1216051422 a.u.
* Info * Gradient : 1.915520e-02 a.u. (RMS)
* Info * 4.090684e-02 a.u. (Max)
* Info * Time : 4.51 sec
Optimization Step 6
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.258837361389 0.932199355478 0.217024126583
C -0.526032242171 0.257236340279 1.282703191145
O -0.296279823532 -0.943367599851 1.238204121665
H -1.405989231958 2.012998583899 0.341286417880
H -2.161642280990 0.405958330882 -0.117178760980
H -0.029612278005 0.884918257485 2.063604407622
H -0.005673662456 -1.225994828731 -0.231881333811
C -0.086437099311 0.467544125948 -1.157261824786
O 0.027133447847 -0.878615377472 -1.167456156214
H 0.846931046732 1.032166568805 -0.964521476681
H -0.597256329041 0.848810120891 -2.056464297712
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.025674740502 0.011350831246 0.033277509296
C -0.000603642568 -0.005348030423 -0.002584001444
O 0.001486988880 0.007408274175 -0.021690363975
H -0.002165360886 0.000777799835 0.002148483854
H 0.000191465214 0.000055699788 -0.001510879457
H 0.001343907750 0.000126555246 -0.000998279033
H -0.001019382509 -0.002820211986 0.006116589993
C 0.027983171276 -0.011498194147 -0.026694720697
O -0.000778501463 0.000637610228 0.014028418675
H 0.000505533882 -0.000469817435 -0.001187437817
H -0.001250906744 -0.000229167302 -0.000851017353
*** Time spent in gradient calculation: 1.73 sec ***
* Info * Energy : -268.1261661882 a.u.
* Info * Gradient : 1.988604e-02 a.u. (RMS)
* Info * 4.353649e-02 a.u. (Max)
* Info * Time : 4.23 sec
Optimization Step 7
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.243810032491 0.925745392870 0.196700768859
C -0.524559037673 0.257786042784 1.284648564835
O -0.297369602509 -0.945213856451 1.248060437769
H -1.391716657173 2.006966751225 0.322481613651
H -2.162982100794 0.407721015070 -0.107740312808
H -0.039378111544 0.885338467745 2.071699248182
H 0.003486732648 -1.214609068551 -0.266904001162
C -0.102393165659 0.475592045181 -1.140580233821
O 0.027358006086 -0.879423421509 -1.174227146753
H 0.834936129724 1.040168461510 -0.957705076254
H -0.589718323450 0.852269138862 -2.055482086239
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.026407093765 0.011999744623 0.033838963804
C -0.002078895913 0.003100416889 -0.003172814046
O 0.002756900853 -0.000515361007 -0.021661409736
H -0.002496771535 0.000717687259 0.002581033795
H -0.000105792286 0.000196838959 -0.001317940499
H 0.001700834916 -0.000454996185 -0.001284377085
H -0.000787172560 0.007467340244 -0.013167348549
C 0.027938928665 -0.012853120928 -0.023575305513
O -0.000573757215 -0.009764386858 0.030782580604
H 0.000814079190 0.000421915091 -0.001585982815
H -0.000741919283 -0.000312570896 -0.001426743784
*** Time spent in gradient calculation: 1.72 sec ***
* Info * Energy : -268.1309409390 a.u.
* Info * Gradient : 2.192561e-02 a.u. (RMS)
* Info * 4.456909e-02 a.u. (Max)
* Info * Time : 4.37 sec
Optimization Step 8
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.232365070120 0.920319042609 0.180513699917
C -0.522189266768 0.254017156881 1.286438058008
O -0.299019407696 -0.943447037865 1.256346661064
H -1.379130050690 2.002204600215 0.306883193990
H -2.164641645604 0.411296159646 -0.100626485781
H -0.047289698114 0.887225072331 2.077006229767
H 0.017188615017 -1.236529993164 -0.271053831859
C -0.114366894701 0.483142421303 -1.127992357492
O 0.027234242866 -0.879131893909 -1.181017938208
H 0.822261666325 1.048739970436 -0.951390932865
H -0.584976860631 0.854479660218 -2.055809188030
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.026722052525 0.012636591568 0.031657055080
C 0.000192328129 -0.004563041876 -0.002252492116
O 0.000936575908 0.006125165830 -0.021211039182
H -0.002736352264 0.000744136740 0.002485119252
H -0.000403278300 0.000488019243 -0.000862490623
H 0.001952175790 -0.000132690900 -0.001203429429
H -0.000337080378 0.001602683803 -0.002467937336
C 0.028388615701 -0.009465019231 -0.021154317269
O -0.000445596895 -0.007610354891 0.019623679781
H -0.000337202318 0.000498981379 -0.002038848976
H -0.000469568520 -0.000325938155 -0.002547786042
*** Time spent in gradient calculation: 1.70 sec ***
* Info * Energy : -268.1350118923 a.u.
* Info * Gradient : 1.957994e-02 a.u. (RMS)
* Info * 4.331190e-02 a.u. (Max)
* Info * Time : 4.25 sec
Optimization Step 9
=====================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.216471998227 0.912597969598 0.160606814827
C -0.520992991445 0.253204240631 1.288105617153
O -0.300149602924 -0.943826148918 1.266507911704
H -1.360374940892 1.995656632016 0.286651180352
H -2.162096581211 0.412416813588 -0.094211907311
H -0.060965891596 0.889224677132 2.085285979061
H 0.026881858755 -1.249143823750 -0.278665700966
C -0.130661197250 0.490510608865 -1.113423952645
O 0.027210377585 -0.876949144432 -1.188690805861
H 0.812966911617 1.048708084270 -0.939726200646
H -0.582733934371 0.860126286471 -2.049031230889
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.025107409705 0.012134103185 0.028255511678
C 0.000378487026 -0.003448828399 -0.001935119535
O 0.000565812432 0.005015585893 -0.020602166677
H -0.002639685029 0.000846009597 0.002559802825
H -0.000607676853 0.000500392962 -0.000427142359
H 0.002194091303 -0.000165556956 -0.001224318674
H 0.000163584660 -0.001511239100 0.003861002372
C 0.023726538604 -0.007084910890 -0.019201665173
O 0.000535501671 -0.005421288389 0.012170104375
H 0.000432316859 0.000179233126 -0.001802482178
H 0.000381506283 -0.001051198347 -0.001615013089
*** Time spent in gradient calculation: 1.68 sec ***
* Info * Energy : -268.1396263809 a.u.
* Info * Gradient : 1.718155e-02 a.u. (RMS)
* Info * 3.969877e-02 a.u. (Max)
* Info * Time : 3.95 sec
Optimization Step 10
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.199317071909 0.903945696897 0.140747327322
C -0.520396857715 0.252849023023 1.289851957571
O -0.300890720113 -0.944584858084 1.277418241949
H -1.339376417524 1.988365162938 0.263430044920
H -2.156199055887 0.411407463035 -0.091506911557
H -0.079026034180 0.891289862708 2.095221966246
H 0.028147253521 -1.250654930999 -0.294904331202
C -0.146371737461 0.496750282369 -1.099592062999
O 0.026635035574 -0.874291184818 -1.196029618265
H 0.801935756100 1.051345025667 -0.927455189411
H -0.584822226477 0.869141948811 -2.040072098582
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.021642456446 0.011218845197 0.023649026183
C 0.000133522970 -0.000508721614 -0.002038315292
O 0.000594121279 0.002284170839 -0.019725842515
H -0.002299173540 0.000849105956 0.002444413676
H -0.000801434081 0.000238423358 -0.000257703813
H 0.002358519951 -0.000327400957 -0.001272063672
H 0.000217007654 0.000654952635 0.000425920469
C 0.018498026188 -0.006356199855 -0.014669609232
O 0.001435896565 -0.007243203978 0.013862367233
H 0.000977092198 0.000383436410 -0.001303485010
H 0.000554786384 -0.001200608219 -0.001079891458
*** Time spent in gradient calculation: 1.76 sec ***
* Info * Energy : -268.1438502193 a.u.
* Info * Gradient : 1.484319e-02 a.u. (RMS)
* Info * 3.396373e-02 a.u. (Max)
* Info * Time : 4.53 sec
Optimization Step 11
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.181150024712 0.894284561742 0.121538745717
C -0.520856704234 0.252843720732 1.291863935858
O -0.301053121643 -0.945716654784 1.289786030267
H -1.316950306639 1.979998129490 0.239195259649
H -2.147550072569 0.409217968994 -0.089925514723
H -0.104374306339 0.894283454576 2.107845339222
H 0.025189465396 -1.253848283784 -0.303177674570
C -0.161374555618 0.502199283561 -1.088218064454
O 0.025395520838 -0.869818676799 -1.204267752480
H 0.793557691665 1.047437209310 -0.916265216197
H -0.589692269518 0.881062986828 -2.029882046567
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.016474901300 0.009125141571 0.016969554465
C -0.000204426442 0.003061693727 -0.002395659767
O 0.000609705502 -0.001219231173 -0.018480406493
H -0.001744131825 0.000715697583 0.002073993584
H -0.000663192405 0.000064316236 0.000062187964
H 0.002400656203 -0.000278230748 -0.001127696946
H 0.000319884886 -0.000412425021 0.003918527307
C 0.011284024337 -0.004782277683 -0.009113103156
O 0.002555931528 -0.004526117536 0.009042306454
H 0.001357839100 -0.000433347734 -0.000741562173
H 0.000585443018 -0.001326005762 -0.000166532550
*** Time spent in gradient calculation: 1.77 sec ***
* Info * Energy : -268.1473560372 a.u.
* Info * Gradient : 1.119650e-02 a.u. (RMS)
* Info * 2.535067e-02 a.u. (Max)
* Info * Time : 4.46 sec
Optimization Step 12
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.164190659990 0.884310199590 0.103967470736
C -0.520729606141 0.248914081329 1.295375250303
O -0.301446544018 -0.944301633731 1.304255333307
H -1.295630440156 1.971286991139 0.214901325474
H -2.139376157856 0.409121776074 -0.092486310384
H -0.134400233507 0.897314963682 2.121641467240
H 0.022833628160 -1.260579687146 -0.329303892518
C -0.172794201983 0.508983106528 -1.079843494420
O 0.022767761287 -0.866534763173 -1.212644744953
H 0.779598254921 1.056892751895 -0.909683351384
H -0.592220393501 0.892451352632 -2.026617469704
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.010694157789 0.006576148000 0.009557919328
C 0.001815558298 -0.002779390067 -0.001949008142
O -0.000950960392 0.004193750173 -0.017098329675
H -0.001233569721 0.000465238515 0.001203838735
H -0.000523296345 0.000162757147 0.000099711860
H 0.002266205653 -0.000159103653 -0.000955487781
H 0.000276374031 0.002839339610 -0.005025941887
C 0.006690751786 -0.002100151458 -0.000988729028
O 0.002400171142 -0.008770689431 0.016913917174
H -0.000222670976 0.000084136888 -0.000522752211
H 0.000200342347 -0.000509347572 -0.001215615418
*** Time spent in gradient calculation: 1.84 sec ***
* Info * Energy : -268.1498371065 a.u.
* Info * Gradient : 9.717029e-03 a.u. (RMS)
* Info * 1.920329e-02 a.u. (Max)
* Info * Time : 4.34 sec
Optimization Step 13
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.150074254402 0.875865899085 0.092358725189
C -0.524397689980 0.250944928069 1.298489878526
O -0.299874392173 -0.947321548671 1.319049643189
H -1.277663609408 1.963962702628 0.197375708101
H -2.130746754721 0.406765309025 -0.092817447681
H -0.168960096348 0.900922581478 2.135827447150
H 0.016950804152 -1.258038097995 -0.323432492966
C -0.179667415396 0.511205519742 -1.078292950900
O 0.019839104827 -0.860757379525 -1.223351230221
H 0.776948560805 1.055630631925 -0.903544538744
H -0.599345073581 0.905036330123 -2.016496036819
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.005223150351 0.003511602772 0.003379244233
C 0.000481266969 0.006663235156 -0.002664021291
O 0.000096886000 -0.005127163059 -0.014838219555
H -0.000604084207 0.000328994158 0.000640903135
H -0.000258358048 0.000171942726 0.000414174567
H 0.001942250605 -0.000158922077 -0.000883608186
H 0.000171909897 -0.002188031932 0.008392457190
C -0.000496087027 -0.001982803737 0.001620092225
O 0.002692707506 -0.000292878981 0.002139290719
H 0.000709025796 0.000151767003 0.000460608659
H 0.000513409915 -0.001093013609 0.001385813648
*** Time spent in gradient calculation: 1.73 sec ***
* Info * Energy : -268.1512776712 a.u.
* Info * Gradient : 6.417602e-03 a.u. (RMS)
* Info * 1.569936e-02 a.u. (Max)
* Info * Time : 4.78 sec
Optimization Step 14
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.141363672170 0.869644364620 0.085999917927
C -0.526082221897 0.246109337282 1.303482769856
O -0.299101897807 -0.945426094347 1.332893729951
H -1.266942445495 1.958334638957 0.186230916965
H -2.124222237785 0.405666403384 -0.100600706184
H -0.200651510005 0.902665015299 2.149191629655
H 0.019026110501 -1.258501456175 -0.346924619919
C -0.181469842981 0.516021609268 -1.081416647302
O 0.016153264104 -0.858431567150 -1.231527331773
H 0.774352606299 1.056833053370 -0.905660109972
H -0.598689838396 0.912951963503 -2.022216149426
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.003419061714 0.001706997184 0.001745711573
C 0.003117871015 -0.001300802766 -0.002136812466
O -0.001760330174 0.002176199138 -0.013347766643
H -0.000196509236 0.000155921213 -0.000266124436
H -0.000033389888 0.000343209055 0.000014433029
H 0.001569849817 0.000008982625 -0.000622921350
H 0.000100606092 0.001553323723 -0.001344339903
C -0.001106344796 0.000324876196 0.003885330557
O 0.002306206346 -0.004335244526 0.011565090609
H -0.000654536745 -0.000182709208 0.000577508710
H 0.000095206100 -0.000455217971 -0.000042006347
*** Time spent in gradient calculation: 1.78 sec ***
* Info * Energy : -268.1523198284 a.u.
* Info * Gradient : 6.047287e-03 a.u. (RMS)
* Info * 1.363809e-02 a.u. (Max)
* Info * Time : 4.13 sec
Optimization Step 15
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.131874821504 0.863642658124 0.079568852421
C -0.532932976780 0.243975845068 1.309946497658
O -0.295849064742 -0.945430277734 1.350928672518
H -1.257770494488 1.951302365853 0.191826996845
H -2.117757360845 0.404133149142 -0.104780307802
H -0.241298243040 0.905188229557 2.164267554164
H 0.021117082285 -1.259937478314 -0.358139326698
C -0.178412573874 0.519027094252 -1.088498659222
O 0.010236099899 -0.855533162315 -1.244385189188
H 0.774281775366 1.065523667159 -0.911902934688
H -0.604497087815 0.924123012622 -2.022008264180
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.001893179740 0.000410274889 -0.001510913059
C 0.002959006411 -0.000266939025 -0.001429676937
O -0.001918826306 0.001370931661 -0.011020558273
H 0.000020029246 -0.000034858365 0.000503322318
H -0.000135378728 0.000334644987 0.000298417941
H 0.001381628552 0.000001263953 -0.000591769860
H 0.000028088398 -0.000102096265 0.001295749347
C 0.000275839592 -0.000411766979 0.003106881188
O 0.001106277811 -0.002405093349 0.007999469703
H -0.001261979595 0.001053010671 0.001000446283
H -0.000542905009 0.000041271267 0.000386909509
*** Time spent in gradient calculation: 1.73 sec ***
* Info * Energy : -268.1533299770 a.u.
* Info * Gradient : 4.606227e-03 a.u. (RMS)
* Info * 1.127005e-02 a.u. (Max)
* Info * Time : 4.14 sec
Optimization Step 16
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.124891177259 0.861284596820 0.078684853975
C -0.538800368732 0.241099793701 1.316336173948
O -0.292749286972 -0.946082718138 1.371483582457
H -1.254516464033 1.950489957969 0.172125438222
H -2.108744504285 0.400362150606 -0.115429765336
H -0.282743265500 0.906713996517 2.178957145356
H 0.023334234926 -1.267092916160 -0.375935296277
C -0.181745439391 0.522399089314 -1.095561976318
O 0.008907858028 -0.850109020313 -1.259680624863
H 0.794714355323 1.030409750580 -0.928077719398
H -0.594041149330 0.933170473013 -2.032247215115
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.001759237700 0.000276915667 0.002445115876
C 0.003103670671 -0.001015384001 -0.003000255163
O -0.001549548701 -0.000106800393 -0.007507771646
H 0.000048628695 0.000326455069 -0.001713039655
H 0.000063892593 0.000180383374 -0.000378502017
H 0.000923805830 0.000066558752 -0.000360845098
H 0.000082085090 -0.002357655840 0.003898746064
C -0.007103537128 0.002136916214 0.002198876059
O 0.003779585064 0.003845479773 0.005035770842
H 0.001650402513 -0.002879702090 -0.000344421123
H 0.000779894758 -0.000485281379 -0.000227811225
*** Time spent in gradient calculation: 1.68 sec ***
* Info * Energy : -268.1540764947 a.u.
* Info * Gradient : 4.660238e-03 a.u. (RMS)
* Info * 7.737035e-03 a.u. (Max)
* Info * Time : 4.34 sec
Optimization Step 17
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.121501862100 0.859721174102 0.079096809416
C -0.544762140767 0.241750806943 1.320777881127
O -0.289934354383 -0.946363829964 1.384512675534
H -1.254049614151 1.948051551887 0.177299834158
H -2.103722316306 0.395074415884 -0.117437834690
H -0.316192279488 0.906605153318 2.191514717978
H 0.023584454117 -1.246876083209 -0.390412782431
C -0.172568685509 0.521043655917 -1.102123290277
O 0.004872921762 -0.849986710505 -1.271838875005
H 0.786211353181 1.058504156511 -0.926911465577
H -0.607817931854 0.940829662051 -2.024968347503
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.003118826210 0.002365221934 0.003824285219
C 0.002687580067 0.003677555089 -0.004771932145
O -0.000855781631 -0.004911426938 -0.004876439785
H -0.000029837787 0.000303519784 -0.001082290509
H -0.000066119031 -0.000190988469 -0.000357829858
H 0.000525395749 -0.000102722965 -0.000125438946
H -0.000788937029 0.004459402734 -0.004717293564
C 0.001733559747 -0.003984899710 0.000156713729
O 0.001495824547 -0.002688835085 0.010463763174
H -0.000597126581 0.000925120150 0.001003264092
H -0.000962682450 0.000145910082 0.000505896090
*** Time spent in gradient calculation: 1.58 sec ***
* Info * Energy : -268.1546438409 a.u.
* Info * Gradient : 5.290527e-03 a.u. (RMS)
* Info * 1.090677e-02 a.u. (Max)
* Info * Time : 3.90 sec
Optimization Step 18
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.115977946780 0.856122234567 0.073562709339
C -0.551266839843 0.238392072749 1.327135018932
O -0.287157251851 -0.942404758522 1.392483344948
H -1.251208984066 1.940501138922 0.208134200537
H -2.103036171097 0.398146434511 -0.115435607380
H -0.335018467333 0.908109863075 2.197948276043
H 0.035071499876 -1.268094333713 -0.397883734820
C -0.171661999133 0.524998241405 -1.104666289090
O 0.001809241077 -0.849909865833 -1.279864626910
H 0.790344758415 1.054464109716 -0.932056733856
H -0.602302667351 0.943768089209 -2.030703218568
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.001126363716 0.000823132239 -0.002922377229
C 0.002458448152 -0.001873607891 -0.000642357303
O -0.002304551858 0.002539469851 -0.005769161567
H -0.000225171966 -0.000240581529 0.001970717386
H -0.000255216135 -0.000050453947 0.000469194319
H 0.001162794204 -0.000060893283 -0.000379149517
H -0.000060301555 -0.002154885704 0.003237141827
C 0.000832470487 -0.000429055357 0.000442182315
O 0.001286702327 0.000989785337 0.003172162627
H -0.000962589500 0.000309840253 0.000530656436
H -0.000783061927 0.000139076353 -0.000071510627
*** Time spent in gradient calculation: 1.72 sec ***
* Info * Energy : -268.1550578552 a.u.
* Info * Gradient : 3.046830e-03 a.u. (RMS)
* Info * 6.711415e-03 a.u. (Max)
* Info * Time : 4.08 sec
Optimization Step 19
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.112444715061 0.854321721283 0.076204370438
C -0.556488013179 0.237869485795 1.332613157294
O -0.283822519648 -0.942527106394 1.402924768208
H -1.249449658444 1.943039192528 0.167894887935
H -2.095793625590 0.394203125330 -0.125802141556
H -0.366426548738 0.907984795380 2.209028931863
H 0.039937139151 -1.262937890782 -0.404995195989
C -0.170316153855 0.526356587803 -1.107550167728
O -0.001301716777 -0.848173896460 -1.290716912487
H 0.802853713057 1.041246621101 -0.940693290886
H -0.595016771026 0.949391091362 -2.032537308841
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.002118424308 0.000299643484 0.004113870509
C 0.002443655840 -0.001280442880 -0.002193756813
O -0.001603842247 -0.000774659413 -0.003835435588
H -0.000049547762 0.000324813684 -0.002208427190
H 0.000029499301 0.000113556100 -0.000304372931
H 0.000714918935 0.000133880026 -0.000360863752
H -0.000095853570 -0.002102004151 0.005388274300
C -0.002251355393 0.001487363874 -0.000525055527
O 0.001749690798 0.002854204014 0.000033602135
H 0.000892267393 -0.000482235615 -0.000291681441
H 0.000317344761 -0.000581110440 0.000221317402
*** Time spent in gradient calculation: 1.71 sec ***
* Info * Energy : -268.1553898659 a.u.
* Info * Gradient : 3.182146e-03 a.u. (RMS)
* Info * 5.784558e-03 a.u. (Max)
* Info * Time : 3.96 sec
Optimization Step 20
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.109874955879 0.852932426239 0.073568300301
C -0.558730871039 0.240804960066 1.330971933747
O -0.281449832262 -0.944000762609 1.414136165541
H -1.238267153156 1.939368603936 0.197363518023
H -2.096925598580 0.401845470624 -0.131351058253
H -0.404032472452 0.908286254779 2.216995685164
H 0.045451421660 -1.234644449344 -0.436194397264
C -0.170080053361 0.524752975848 -1.106842659474
O -0.003777115535 -0.849433682428 -1.299988975458
H 0.805094009086 1.039570141873 -0.939754454801
H -0.584374847053 0.951970311973 -2.033899079519
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.002654244702 -0.000038740926 0.002315195468
C 0.003006272170 0.011257687540 -0.006770807055
O -0.000458761821 -0.010992864751 -0.001188104509
H 0.000215954965 -0.000091850683 0.001019550390
H 0.000092359699 0.000103064990 -0.000973497227
H -0.000187936409 -0.000006299954 0.000636577052
H -0.002275761510 0.014564604881 -0.022481744688
C -0.004278063779 -0.002535805331 0.003965071991
O 0.003976229007 -0.011851509693 0.023877232507
H 0.001512315179 0.000597423423 -0.000583000329
H 0.001070063910 -0.000989581926 0.000160064535
*** Time spent in gradient calculation: 1.64 sec ***
* Info * Energy : -268.1548739649 a.u.
* Info * Gradient : 1.284065e-02 a.u. (RMS)
* Info * 2.695164e-02 a.u. (Max)
* Info * Time : 3.86 sec
Optimization Step 21
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.111933937669 0.854216603476 0.075374715692
C -0.557112540706 0.238025343356 1.333149961170
O -0.283490123324 -0.942305228237 1.403774679620
H -1.249653177647 1.942195951873 0.173787185510
H -2.095715657500 0.394084203674 -0.125182913318
H -0.368819354019 0.907749236824 2.210276744124
H 0.040002907312 -1.261503693549 -0.411458744500
C -0.169836542617 0.526101383055 -1.107616292045
O -0.001638606445 -0.848416933252 -1.291414195930
H 0.801277734081 1.042449942966 -0.939875111390
H -0.595329479311 0.950700665578 -2.032919203451
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.001884509940 0.000543814879 0.002945105808
C 0.002254112721 -0.000467741671 -0.001968992445
O -0.001519898808 -0.001160585948 -0.003799253215
H -0.000114683296 0.000218490264 -0.001523553297
H -0.000012809278 0.000035859925 -0.000224217351
H 0.000783851001 0.000051845278 -0.000301516230
H -0.000362367576 0.000208943549 0.000604135031
C -0.001179356157 0.000748684010 0.000237702522
O 0.001881422600 0.000410761138 0.004614367963
H 0.000228777354 -0.000414381932 -0.000246760903
H -0.000047225494 -0.000178364366 -0.000311436010
*** Time spent in gradient calculation: 1.62 sec ***
* Info * Energy : -268.1554868959 a.u.
* Info * Gradient : 2.538708e-03 a.u. (RMS)
* Info * 5.000087e-03 a.u. (Max)
* Info * Time : 4.21 sec
Optimization Step 22
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.110902267066 0.853809514622 0.074125657418
C -0.558445121706 0.238137702446 1.334212042649
O -0.282761842503 -0.941769266752 1.405574590493
H -1.248655625052 1.940937625892 0.181655574559
H -2.095449810518 0.394127070962 -0.124354502616
H -0.374829546068 0.907644351458 2.212465205848
H 0.041995088271 -1.262875003112 -0.414681231648
C -0.169200333387 0.525877202258 -1.107796479597
O -0.002450101623 -0.848557844114 -1.293591928800
H 0.800641940415 1.043908123763 -0.938390280411
H -0.594830574729 0.951919212831 -2.031867227458
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.001580734350 0.000672806850 0.001207025548
C 0.002153380700 0.000264432529 -0.001489189068
O -0.001552821953 -0.001341397925 -0.003749302350
H -0.000123897318 0.000118572642 -0.000623621170
H -0.000037170765 -0.000067196303 -0.000078937165
H 0.000827119952 -0.000013804635 -0.000276793036
H -0.000356108313 0.000222712221 0.000319128982
C -0.000974302517 0.000250820341 0.000171743816
O 0.001751887968 0.000182978887 0.004642560259
H -0.000019244943 -0.000094635472 -0.000103866410
H -0.000060725369 -0.000197417615 0.000005578009
*** Time spent in gradient calculation: 1.55 sec ***
* Info * Energy : -268.1555780090 a.u.
* Info * Gradient : 2.272982e-03 a.u. (RMS)
* Info * 4.965477e-03 a.u. (Max)
* Info * Time : 3.52 sec
Optimization Step 23
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.109393775737 0.853139217244 0.073227223600
C -0.560629880362 0.237783224081 1.335721882502
O -0.281491587207 -0.940820401948 1.408523747941
H -1.247108135481 1.939760517491 0.186385387376
H -2.094868270527 0.394837450676 -0.124203099796
H -0.385066838346 0.907634486021 2.215579093197
H 0.045267811521 -1.264216025843 -0.418948193535
C -0.168258928598 0.525816529247 -1.108076628994
O -0.003772110795 -0.848686568915 -1.297111563421
H 0.801383645279 1.043758945582 -0.937454568585
H -0.593446369875 0.953861894584 -2.031704387655
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.001291901987 0.000636051671 0.000230580554
C 0.002168652130 0.000046920181 -0.001297586569
O -0.001728539376 -0.000856276620 -0.003558168852
H -0.000160060968 0.000059236720 -0.000101176671
H -0.000035833673 -0.000079845675 0.000043256429
H 0.000842679187 0.000006036896 -0.000228311915
H -0.000332314713 0.000148875353 0.000383828466
C -0.000770779373 -0.000048469100 0.000501660798
O 0.001644251742 0.000102456650 0.004199316862
H -0.000147519349 0.000117535492 -0.000050125777
H -0.000161427158 -0.000133562936 -0.000100416872
*** Time spent in gradient calculation: 1.91 sec ***
* Info * Energy : -268.1556863787 a.u.
* Info * Gradient : 2.073139e-03 a.u. (RMS)
* Info * 4.510912e-03 a.u. (Max)
* Info * Time : 3.97 sec
Optimization Step 24
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.107431088694 0.852198939806 0.072592034150
C -0.563941376117 0.237481416671 1.337909308420
O -0.279396615797 -0.939785867329 1.412744083437
H -1.244469244771 1.938697019058 0.188277569931
H -2.093880826736 0.395954757443 -0.125291760878
H -0.400635186744 0.907479338312 2.220072583659
H 0.049887178496 -1.265560108275 -0.425037135585
C -0.166989357906 0.525906621094 -1.108782153128
O -0.005715565389 -0.848763361849 -1.302039278482
H 0.804066958571 1.040751668917 -0.936428493747
H -0.590745169990 0.956866797680 -2.031190220004
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.001112625501 0.000434600409 -0.000044403249
C 0.002043547534 0.000511930937 -0.001255180769
O -0.001731637111 -0.001146820837 -0.003168528483
H -0.000138722479 0.000019617025 0.000109978528
H -0.000039660102 -0.000075214065 0.000085192046
H 0.000806935946 0.000020672524 -0.000219343658
H -0.000305434280 0.000157811946 0.000288733909
C -0.000991292528 0.000147771485 0.000225783657
O 0.001666792552 0.000045560742 0.003921011539
H -0.000077959841 0.000116809743 -0.000019640435
H -0.000093081017 -0.000232935164 0.000097678698
*** Time spent in gradient calculation: 1.66 sec ***
* Info * Energy : -268.1558290942 a.u.
* Info * Gradient : 1.955084e-03 a.u. (RMS)
* Info * 4.260822e-03 a.u. (Max)
* Info * Time : 3.84 sec
Optimization Step 25
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.104699753113 0.851069018850 0.071970306814
C -0.568829510300 0.236459996046 1.340945100586
O -0.276198472832 -0.937908641685 1.418335931804
H -1.240929490903 1.937764402163 0.187568390561
H -2.092228605463 0.397488532527 -0.127492249897
H -0.422851999477 0.906472920391 2.226325621711
H 0.056513900852 -1.266818337590 -0.433119805096
C -0.164856222953 0.525883393000 -1.109294542015
O -0.008741732191 -0.848977635257 -1.308867465726
H 0.807360267867 1.037447152342 -0.935451453976
H -0.586869399469 0.961323670472 -2.030963162696
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000872867412 0.000289957873 0.000056943915
C 0.002107062060 -0.000407358355 -0.001284284470
O -0.001979689892 -0.000268348104 -0.002643460824
H -0.000132967110 0.000026782742 0.000034723016
H -0.000039512780 -0.000066999816 0.000102613535
H 0.000787198917 0.000025380345 -0.000220061103
H -0.000274450525 0.000252063221 0.000312755032
C -0.000676371728 0.000182848909 0.000663024218
O 0.001541831815 -0.000101336820 0.003283188858
H -0.000247675449 0.000192736207 -0.000093684961
H -0.000187883914 -0.000124658209 -0.000192417715
*** Time spent in gradient calculation: 1.62 sec ***
* Info * Energy : -268.1560154692 a.u.
* Info * Gradient : 1.740361e-03 a.u. (RMS)
* Info * 3.628615e-03 a.u. (Max)
* Info * Time : 3.79 sec
Optimization Step 26
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.101179074827 0.849748821732 0.070978514820
C -0.576065504102 0.236129649058 1.345410538201
O -0.271038904289 -0.935956956608 1.425321136123
H -1.236092369871 1.936542880601 0.188349317892
H -2.090280934444 0.400153474819 -0.130744086269
H -0.456212028327 0.904685957027 2.235786905227
H 0.066340171663 -1.269871469070 -0.444532313195
C -0.162016564789 0.525664352629 -1.110455111885
O -0.013102455259 -0.849153899925 -1.317652149360
H 0.814529965446 1.028349701091 -0.932464358818
H -0.580202211631 0.968098539116 -2.029315836355
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000775684899 -0.000099233508 -0.000111046794
C 0.001689609588 0.001619457356 -0.001080518748
O -0.001668744662 -0.002116160881 -0.001994181207
H -0.000097699441 -0.000013997979 0.000168289146
H -0.000037597229 -0.000045815836 0.000108211574
H 0.000701174633 0.000074262818 -0.000188413103
H -0.000154464933 0.000087685778 0.000287603612
C -0.001431906096 0.000585729491 -0.000410306395
O 0.001668868052 0.000188779913 0.002951270530
H 0.000168754279 -0.000007487313 -0.000025654122
H -0.000041055632 -0.000273227660 0.000314611638
*** Time spent in gradient calculation: 1.60 sec ***
* Info * Energy : -268.1562504621 a.u.
* Info * Gradient : 1.745271e-03 a.u. (RMS)
* Info * 3.395697e-03 a.u. (Max)
* Info * Time : 3.77 sec
Optimization Step 27
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.096661182652 0.848540999530 0.069869648441
C -0.586016796409 0.233590796742 1.350667305704
O -0.263689005004 -0.931590873289 1.433528591737
H -1.230601555844 1.935644543099 0.186613485891
H -2.087109980359 0.403403736757 -0.136540519419
H -0.499853427133 0.899604896977 2.247200147316
H 0.077793980161 -1.272289310833 -0.458879342188
C -0.157074694209 0.524842815166 -1.110095739535
O -0.019745468285 -0.849917656310 -1.329011301893
H 0.820778563834 1.022054055901 -0.928040392585
H -0.570173561092 0.975901704980 -2.028207426220
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000429760405 0.000014903782 0.000044494608
C 0.002164931766 -0.001171493944 -0.000937128571
O -0.002387250237 0.000536370135 -0.001290430297
H -0.000161060565 0.000000317590 -0.000018504369
H 0.000009719774 -0.000065572100 0.000045470484
H 0.000734674750 -0.000000960298 -0.000168881792
H -0.000112040527 0.000434768845 0.000215510332
C -0.000464951975 0.000160263941 0.000615780054
O 0.001225747697 -0.000150597273 0.001927733194
H -0.000380352929 0.000331928115 -0.000149364247
H -0.000186197104 -0.000091664063 -0.000264686446
*** Time spent in gradient calculation: 1.62 sec ***
* Info * Energy : -268.1565274206 a.u.
* Info * Gradient : 1.410286e-03 a.u. (RMS)
* Info * 2.766201e-03 a.u. (Max)
* Info * Time : 3.82 sec
Optimization Step 28
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.091736216392 0.847229173159 0.068160345985
C -0.599195855034 0.232299623317 1.357001086225
O -0.252857370745 -0.927043452289 1.442121236602
H -1.223337682760 1.934467771824 0.188243302008
H -2.084844680268 0.409078141850 -0.142260731358
H -0.558947801577 0.892493339088 2.261099137466
H 0.090714394950 -1.278419577071 -0.476823335432
C -0.150964942059 0.523745108108 -1.109964689822
O -0.028255743456 -0.850623841610 -1.341566288219
H 0.833714819823 1.004819696959 -0.919275258080
H -0.554751423278 0.987746692620 -2.024798822830
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000423685101 -0.000463491494 -0.000451183651
C 0.001656025209 0.001344521065 -0.000506831039
O -0.001953346258 -0.001726008410 -0.000592735139
H -0.000167311568 -0.000065305808 0.000208831345
H -0.000023393758 -0.000055040258 0.000132873449
H 0.000632286753 0.000071322238 -0.000105777055
H 0.000067635256 0.000173108700 0.000222392738
C -0.001071337382 0.000518383702 -0.000459550183
O 0.001288339882 0.000288459735 0.001545527455
H 0.000109887031 0.000036855077 -0.000109690834
H -0.000106851038 -0.000130260207 0.000139956500
*** Time spent in gradient calculation: 1.76 sec ***
* Info * Energy : -268.1568371473 a.u.
* Info * Gradient : 1.313596e-03 a.u. (RMS)
* Info * 2.673201e-03 a.u. (Max)
* Info * Time : 4.10 sec
Optimization Step 29
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.086770465124 0.846985720011 0.066981682235
C -0.613401264334 0.229036758737 1.362282171435
O -0.240888621299 -0.920995956210 1.449049036029
H -1.214684429937 1.934762724956 0.188041766381
H -2.082184334685 0.416950523242 -0.150804190627
H -0.620776515360 0.880999362702 2.273419799091
H 0.100393970616 -1.283588223343 -0.493403506891
C -0.144373061397 0.521948001028 -1.108227058877
O -0.037460667411 -0.851909537117 -1.353097619276
H 0.844066215655 0.990784009440 -0.908723694960
H -0.537736241571 0.997499054426 -2.021785829145
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000209560330 -0.000492986590 -0.000208285652
C 0.001975925914 0.000270442062 -0.000315774606
O -0.002337127184 -0.000632235011 -0.000028874713
H -0.000208362256 -0.000050755345 0.000113235027
H 0.000063698128 -0.000072339387 -0.000037845952
H 0.000613948667 0.000040945104 -0.000085843583
H 0.000122853534 0.000358844932 0.000153604230
C -0.000806266782 0.000341815803 -0.000273111003
O 0.001012663871 0.000202354380 0.000806152767
H -0.000137549192 0.000177627979 -0.000075763812
H -0.000083403331 -0.000153055143 -0.000019623434
*** Time spent in gradient calculation: 1.64 sec ***
* Info * Energy : -268.1571226699 a.u.
* Info * Gradient : 1.109379e-03 a.u. (RMS)
* Info * 2.421305e-03 a.u. (Max)
* Info * Time : 4.06 sec
Optimization Step 30
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.082234991154 0.847610695782 0.065845011076
C -0.628822413186 0.225547089168 1.366702132182
O -0.227324260851 -0.914466789166 1.453655354160
H -1.203156854947 1.936215525113 0.188366987156
H -2.082080103529 0.428325405319 -0.155062716942
H -0.686679992692 0.866085848655 2.284323439763
H 0.105300560157 -1.291373014893 -0.508861829408
C -0.137484263008 0.519700657518 -1.105673683800
O -0.046618443244 -0.853517236935 -1.362767724113
H 0.855932323151 0.974192743630 -0.898289829203
H -0.520936443836 1.007896777250 -2.016881144315
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000357239083 -0.000615872884 -0.000420252192
C 0.001835292348 0.000340773764 -0.000134200059
O -0.002306984578 -0.000712927271 0.000312804069
H -0.000165231449 -0.000069847416 0.000003280562
H -0.000042829555 -0.000130803418 0.000203807970
H 0.000584554579 0.000115211723 -0.000076773460
H 0.000123690225 0.000208482306 0.000011730886
C -0.000368181696 0.000443529673 -0.000299735024
O 0.000952311935 0.000248040721 0.000605620123
H -0.000040297263 0.000149593734 -0.000188751190
H -0.000207179963 0.000017936120 0.000018094218
*** Time spent in gradient calculation: 1.63 sec ***
* Info * Energy : -268.1574008819 a.u.
* Info * Gradient : 1.065458e-03 a.u. (RMS)
* Info * 2.434808e-03 a.u. (Max)
* Info * Time : 4.06 sec
Optimization Step 31
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.077631531683 0.850210794140 0.065526802587
C -0.645196616473 0.221124893340 1.369744455994
O -0.212281347739 -0.907619123784 1.455519589367
H -1.190182471740 1.939574099619 0.191674397461
H -2.081183511955 0.444348195522 -0.164900260584
H -0.756761475641 0.845543972253 2.293595476589
H 0.106527400496 -1.298239420567 -0.520238866406
C -0.131597604454 0.516802053252 -1.102546957523
O -0.055720229679 -0.855606272317 -1.369855857562
H 0.866100183863 0.956549871768 -0.884555247277
H -0.500968952025 1.014609431739 -2.014185330158
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000157882785 -0.000542275917 0.000322750818
C 0.001797217720 0.001045714148 -0.000127706654
O -0.002098463910 -0.001277866408 0.000555834297
H -0.000229244685 -0.000037483707 0.000030542210
H 0.000131274247 -0.000122424544 -0.000189730728
H 0.000489600464 0.000156560662 -0.000046232007
H 0.000085802513 0.000082518677 0.000146493202
C -0.000969674008 0.000225812297 -0.000518800150
O 0.001088189738 0.000516609782 0.000002714642
H -0.000100032966 0.000120917788 -0.000017201553
H -0.000032158732 -0.000168461366 -0.000114142187
*** Time spent in gradient calculation: 1.66 sec ***
* Info * Energy : -268.1576836936 a.u.
* Info * Gradient : 1.140668e-03 a.u. (RMS)
* Info * 2.519017e-03 a.u. (Max)
* Info * Time : 4.25 sec
Optimization Step 32
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.074185597638 0.853452539982 0.065122728661
C -0.661656647090 0.215651802502 1.371186298118
O -0.196911381686 -0.900075511682 1.454766026075
H -1.173655099865 1.943881577068 0.194448623590
H -2.083829516585 0.463084476309 -0.168031362614
H -0.823423412025 0.822118020068 2.299761772416
H 0.101515630066 -1.307223061528 -0.529169859267
C -0.125138320735 0.513628468367 -1.098329661003
O -0.064243446670 -0.858076840790 -1.374400643644
H 0.876343154661 0.940183216518 -0.872256817177
H -0.484023953114 1.022390013635 -2.008231147452
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000574231925 -0.000267082698 0.000371467662
C 0.001652901890 0.000520904079 -0.000184727569
O -0.002023744319 -0.000842388031 0.000539644586
H -0.000094471369 -0.000048832449 -0.000124208430
H -0.000023967601 -0.000243139597 0.000095168856
H 0.000499209511 0.000245205932 -0.000060549220
H -0.000115798915 -0.000217621015 -0.000042085226
C -0.000305800239 0.000141228759 -0.000637461085
O 0.001272931380 0.000485580534 0.000187690049
H -0.000078314017 0.000150761537 -0.000118488333
H -0.000210967707 0.000077074564 0.000018606583
*** Time spent in gradient calculation: 1.65 sec ***
* Info * Energy : -268.1579603190 a.u.
* Info * Gradient : 1.029073e-03 a.u. (RMS)
* Info * 2.257515e-03 a.u. (Max)
* Info * Time : 4.09 sec
Optimization Step 33
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.070743962718 0.857271600158 0.065065689816
C -0.677570745267 0.209961356852 1.371938564394
O -0.181488666945 -0.892361200947 1.452928460257
H -1.159629796826 1.948116774098 0.200801542682
H -2.085067343466 0.483495013113 -0.176032588683
H -0.890114763943 0.794306409840 2.304496498095
H 0.097659994371 -1.314412208961 -0.535763316923
C -0.119244176329 0.510344988130 -1.094137851276
O -0.072990768705 -0.860567980952 -1.377483392812
H 0.885068527473 0.924080199513 -0.857544831331
H -0.463512729873 1.027368451366 -2.005302303262
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000557044165 -0.000177916833 0.000846160602
C 0.001795162297 0.001003459641 -0.000075564298
O -0.001913423902 -0.000950692935 0.000426788609
H -0.000235669738 -0.000068959593 0.000014024848
H 0.000044255381 -0.000144564844 -0.000144787539
H 0.000407370822 0.000153542781 0.000019692077
H -0.000252316708 -0.000416599321 -0.000002322930
C -0.000376095549 -0.000363451866 -0.000787220826
O 0.001396958439 0.000589549433 -0.000028137835
H -0.000218421133 0.000293630117 0.000006646023
H -0.000095645319 0.000082974083 -0.000237214435
*** Time spent in gradient calculation: 1.69 sec ***
* Info * Energy : -268.1582251202 a.u.
* Info * Gradient : 1.126991e-03 a.u. (RMS)
* Info * 2.178797e-03 a.u. (Max)
* Info * Time : 3.92 sec
Optimization Step 34
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.068327842437 0.860503895802 0.064848347576
C -0.691069299111 0.204377340028 1.371777968457
O -0.168405068774 -0.885416454846 1.451682151968
H -1.145328654092 1.951906910730 0.205303337435
H -2.086866966327 0.501355925870 -0.181719969506
H -0.943388363353 0.770785988863 2.305616000191
H 0.095047256807 -1.321171139412 -0.541964987162
C -0.113932481030 0.507623437710 -1.090141196546
O -0.080447865131 -0.862529574439 -1.379880399355
H 0.893060498540 0.909328572087 -0.845378622917
H -0.447970004846 1.032811832279 -2.000473547146
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000901400663 0.000115947317 0.001015048611
C 0.001747618242 0.000736685938 -0.000039801426
O -0.001773458347 -0.000754662466 0.000335444024
H -0.000169424544 -0.000038377035 -0.000038881191
H 0.000015480323 -0.000170595780 -0.000145095479
H 0.000400770969 0.000197548499 0.000018455121
H -0.000365980159 -0.000616214765 -0.000088447512
C -0.000012360921 -0.000513441073 -0.001009281585
O 0.001540084169 0.000562411903 0.000110627661
H -0.000266268852 0.000262359115 0.000034983182
H -0.000218772199 0.000219654446 -0.000159338593
*** Time spent in gradient calculation: 1.51 sec ***
* Info * Energy : -268.1584375794 a.u.
* Info * Gradient : 1.141402e-03 a.u. (RMS)
* Info * 1.956321e-03 a.u. (Max)
* Info * Time : 3.78 sec
Optimization Step 35
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.065881577765 0.862976737922 0.064708080350
C -0.702739779236 0.199473748200 1.371683886551
O -0.157258663670 -0.879098516509 1.450932330952
H -1.133918552508 1.954602458689 0.209766870201
H -2.087856768585 0.516660265214 -0.186542416055
H -0.990234505739 0.748437490121 2.305837337916
H 0.096382338210 -1.326347493844 -0.548125923571
C -0.109348972325 0.505331505032 -1.086750391713
O -0.087334874367 -0.864078792831 -1.382128095567
H 0.899854936801 0.896693550273 -0.834753206678
H -0.432475652072 1.036413087855 -1.997722036484
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000923307964 0.000102360667 0.001134945830
C 0.001621612279 0.000981292731 0.000155750330
O -0.001631825104 -0.000836592854 0.000184831913
H -0.000209018760 -0.000050003193 -0.000041740297
H 0.000018620222 -0.000118484450 -0.000144601305
H 0.000370910329 0.000156134203 0.000070287722
H -0.000365488536 -0.000705607630 -0.000116025422
C 0.000036052650 -0.000630808512 -0.001023366633
O 0.001493348687 0.000561455227 0.000059651567
H -0.000284107620 0.000332192407 0.000059722119
H -0.000130211352 0.000209570865 -0.000306119044
*** Time spent in gradient calculation: 1.60 sec ***
* Info * Energy : -268.1586160979 a.u.
* Info * Gradient : 1.144332e-03 a.u. (RMS)
* Info * 1.901794e-03 a.u. (Max)
* Info * Time : 3.83 sec
Optimization Step 36
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.063522142653 0.865456024134 0.064466110704
C -0.713626156577 0.194247757306 1.370686343706
O -0.146889860092 -0.873065033670 1.450455896327
H -1.122179852362 1.957258134983 0.214271615790
H -2.088327665538 0.531772262208 -0.192598316026
H -1.032669045372 0.727432266021 2.303784693088
H 0.100869236418 -1.330210650438 -0.553885781846
C -0.105230755791 0.503213357055 -1.083221580617
O -0.093936627332 -0.865417509742 -1.383778069450
H 0.906611608477 0.883882174707 -0.825820338871
H -0.419927581019 1.040376361094 -1.993346539095
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000976472221 0.000225901672 0.001191283104
C 0.001569861903 0.000799780008 0.000124503685
O -0.001555129159 -0.000711036178 0.000115712936
H -0.000216678330 -0.000008418934 -0.000075757545
H 0.000043789644 -0.000085100720 -0.000218468992
H 0.000367515303 0.000186384563 0.000033609106
H -0.000289275044 -0.000674964788 -0.000133666723
C -0.000048929091 -0.000742768476 -0.000929819106
O 0.001474293145 0.000519235142 0.000039359381
H -0.000206918734 0.000262231981 0.000039236379
H -0.000167338638 0.000228884734 -0.000151140691
*** Time spent in gradient calculation: 1.51 sec ***
* Info * Energy : -268.1587785304 a.u.
* Info * Gradient : 1.103635e-03 a.u. (RMS)
* Info * 1.766243e-03 a.u. (Max)
* Info * Time : 3.78 sec
Optimization Step 37
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.060323819422 0.867720015376 0.064054313342
C -0.725195609845 0.188473778735 1.369551580103
O -0.136358800843 -0.866759773939 1.449901975506
H -1.108427559629 1.959587982248 0.218808415503
H -2.088795136340 0.547273124767 -0.195754582201
H -1.076661055102 0.703862993102 2.301071167086
H 0.109143489835 -1.332621410249 -0.559493762947
C -0.101193575446 0.501504766221 -1.080253386988
O -0.101031807361 -0.866664588859 -1.384735857394
H 0.912906237877 0.871936491495 -0.817540925220
H -0.406490945595 1.042960683828 -1.990893886315
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000653700242 -0.000002589550 0.000979019124
C 0.000955040050 0.000923649739 0.000260124447
O -0.001345907722 -0.000765327208 -0.000075628114
H -0.000136002113 -0.000016609047 -0.000166659073
H 0.000014923618 -0.000135174797 -0.000020381461
H 0.000433079572 0.000190896979 0.000097126976
H -0.000123463922 -0.000534557605 -0.000167149470
C -0.000084094395 -0.000473601830 -0.000765328107
O 0.001200516781 0.000448788225 0.000048416758
H -0.000210123396 0.000237535659 0.000039082618
H -0.000059460534 0.000123428745 -0.000192583705
*** Time spent in gradient calculation: 1.64 sec ***
* Info * Energy : -268.1589400326 a.u.
* Info * Gradient : 8.979521e-04 a.u. (RMS)
* Info * 1.550133e-03 a.u. (Max)
* Info * Time : 3.94 sec
Optimization Step 38
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.056721621053 0.870718886127 0.063238987784
C -0.737215421264 0.181215464304 1.366770414893
O -0.124800606847 -0.859987359604 1.449710194715
H -1.094022853487 1.962149460825 0.226283933581
H -2.088430218045 0.566285760390 -0.203402841853
H -1.126344940094 0.676278709807 2.294326206092
H 0.118833827691 -1.333138633036 -0.563889078229
C -0.097017839142 0.499888524569 -1.076762377021
O -0.109069001853 -0.867898195966 -1.384652114346
H 0.920768380267 0.858226852834 -0.811363416887
H -0.394642820556 1.046669860638 -1.986361212029
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000468970994 -0.000018429875 0.000634304984
C 0.001207734878 0.000674264560 0.000163109673
O -0.001407191375 -0.000599326025 -0.000104368254
H -0.000221557188 0.000004711257 -0.000114648614
H -0.000021088889 -0.000050577957 -0.000129213187
H 0.000335634166 0.000202441039 -0.000020150584
H 0.000085090879 -0.000195373006 -0.000079201452
C -0.000447155702 -0.000541863759 -0.000222113818
O 0.000990550645 0.000303018779 -0.000093656906
H 0.000042294249 0.000098924777 -0.000083290115
H -0.000106972215 0.000114112022 0.000084816096
*** Time spent in gradient calculation: 1.66 sec ***
* Info * Energy : -268.1591028229 a.u.
* Info * Gradient : 7.905853e-04 a.u. (RMS)
* Info * 1.533060e-03 a.u. (Max)
* Info * Time : 3.94 sec
Optimization Step 39
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.051675045160 0.874036892866 0.062393634700
C -0.751692500423 0.172039326515 1.363656575954
O -0.111499191056 -0.852224133938 1.450110445804
H -1.072944500877 1.964755028485 0.234836570441
H -2.088121750513 0.588290892380 -0.206847406679
H -1.182138693267 0.641752115302 2.286313120337
H 0.122790956670 -1.335024548853 -0.567412187860
C -0.092589836127 0.498965024763 -1.075082281467
O -0.117779388645 -0.869004946489 -1.383879422955
H 0.926849971474 0.847006722218 -0.804155300236
H -0.380661085400 1.048257090788 -1.986307979037
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000089042339 -0.000273379287 0.000330165379
C 0.000174652228 0.000868023687 0.000201796244
O -0.000905102668 -0.000821291845 -0.000135260470
H -0.000022650122 -0.000020195231 -0.000193857456
H -0.000018343277 -0.000207529686 0.000166552231
H 0.000378306345 0.000165076116 0.000140653632
H 0.000127391076 -0.000121413185 -0.000101675342
C -0.000269855414 0.000053311294 -0.000394100395
O 0.000604694508 0.000214407260 0.000116142371
H -0.000178179101 0.000144567682 0.000041612631
H 0.000006662350 -0.000012773807 -0.000138802921
*** Time spent in gradient calculation: 1.85 sec ***
* Info * Energy : -268.1592621729 a.u.
* Info * Gradient : 5.720331e-04 a.u. (RMS)
* Info * 1.229645e-03 a.u. (Max)
* Info * Time : 4.04 sec
Optimization Step 40
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.048617476062 0.877574691313 0.061437432395
C -0.763710079487 0.163066542284 1.359264480978
O -0.098928676958 -0.844419755153 1.450534760876
H -1.057471415259 1.966909400694 0.245819593092
H -2.087782475129 0.610748707157 -0.216504918666
H -1.234426455935 0.608425432012 2.274330277044
H 0.122675784618 -1.336744318688 -0.569834416110
C -0.087784055166 0.497483086486 -1.071463851684
O -0.126262205232 -0.870031205029 -1.383309540309
H 0.935316462660 0.832472393879 -0.797620662051
H -0.367435383631 1.052072221138 -1.981833289017
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000224050982 -0.000008994456 0.000055642681
C 0.001132982239 0.000307367167 0.000094555534
O -0.001075594229 -0.000466525249 -0.000037182754
H -0.000180276329 -0.000000577319 -0.000018496865
H -0.000065987349 -0.000024894504 -0.000044828918
H 0.000117912584 0.000145133191 -0.000072153034
H 0.000121351617 0.000117656605 0.000057441123
C -0.000436112875 -0.000166829764 0.000093787926
O 0.000554145664 0.000058763756 -0.000068873686
H 0.000132418117 0.000007119616 -0.000133496758
H -0.000091919653 0.000019963243 0.000105761981
*** Time spent in gradient calculation: 1.92 sec ***
* Info * Energy : -268.1593859022 a.u.
* Info * Gradient : 5.661493e-04 a.u. (RMS)
* Info * 1.177737e-03 a.u. (Max)
* Info * Time : 4.34 sec
Optimization Step 41
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.044768720860 0.879903967640 0.060570501820
C -0.776451912135 0.153964227246 1.355625483978
O -0.086478547930 -0.836221553012 1.451883551826
H -1.038563645792 1.967880330812 0.254727075243
H -2.086835520835 0.630296795933 -0.221787199444
H -1.281279327331 0.575151981507 2.264095680597
H 0.119127080308 -1.340443694463 -0.573661369922
C -0.082966211323 0.496506331494 -1.069297611776
O -0.134533572538 -0.870542715027 -1.383658480469
H 0.941102310921 0.820728189833 -0.788129002071
H -0.352288658648 1.054855171662 -1.980564147618
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000150515845 -0.000167127053 -0.000199254229
C 0.000099614837 0.000438549274 0.000066735781
O -0.000457213986 -0.000620914871 0.000028426431
H 0.000031349004 -0.000005282893 -0.000055044360
H 0.000002193355 -0.000160820340 0.000155442964
H 0.000169584301 0.000079676808 0.000108203390
H 0.000022274413 0.000096393025 0.000067168772
C -0.000172468368 0.000320863680 -0.000180612615
O 0.000281498550 -0.000014376684 0.000104620831
H -0.000122315447 0.000068773207 0.000018904925
H -0.000024546107 -0.000048680108 -0.000083420725
*** Time spent in gradient calculation: 1.74 sec ***
* Info * Energy : -268.1594761510 a.u.
* Info * Gradient : 3.428832e-04 a.u. (RMS)
* Info * 7.716139e-04 a.u. (Max)
* Info * Time : 4.29 sec
Optimization Step 42
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.042619512345 0.881817068486 0.059771032223
C -0.787000908051 0.145690183335 1.351659899047
O -0.075032183324 -0.827809870338 1.453230631630
H -1.025656739802 1.968121589122 0.264253965477
H -2.086079504106 0.648396698034 -0.230698143146
H -1.324205029486 0.544494573607 2.251638033312
H 0.116786519663 -1.344504460130 -0.579443039346
C -0.077619665955 0.494800775091 -1.065169097410
O -0.142720859279 -0.870863621724 -1.385204209762
H 0.948973045597 0.805324589062 -0.777969312968
H -0.336004372346 1.059248152155 -1.975604061818
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000045269874 -0.000072040283 -0.000391043349
C 0.000608685715 0.000024250572 0.000094026683
O -0.000477794242 -0.000279647651 0.000033767665
H -0.000060715266 -0.000013696996 0.000062131156
H -0.000046086903 -0.000016951325 0.000046609461
H 0.000006878102 0.000065826814 -0.000047723325
H -0.000006648378 0.000267500116 0.000134821137
C -0.000210607633 0.000234111299 0.000161599914
O 0.000162404038 -0.000112884257 -0.000034596671
H 0.000084003651 -0.000018295668 -0.000076939324
H -0.000038247246 -0.000092602566 0.000044976670
*** Time spent in gradient calculation: 1.73 sec ***
* Info * Energy : -268.1595330501 a.u.
* Info * Gradient : 3.229734e-04 a.u. (RMS)
* Info * 6.163825e-04 a.u. (Max)
* Info * Time : 4.05 sec
Optimization Step 43
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.040596855610 0.882461645574 0.059508445626
C -0.794227289407 0.140345205556 1.349576510414
O -0.067580403904 -0.821927822979 1.454939077624
H -1.015769413787 1.967804746089 0.269336254653
H -2.084698824191 0.658681638396 -0.235546711220
H -1.350116934096 0.524570419487 2.244591482597
H 0.115029827748 -1.348923249663 -0.585533895499
C -0.074114991082 0.493727521115 -1.063101448618
O -0.147865898422 -0.870740896759 -1.387343016338
H 0.953128144759 0.796185404967 -0.770695884660
H -0.325498302861 1.062597086937 -1.972782666267
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000146637413 -0.000159551419 -0.000376098475
C 0.000056284310 0.000103080170 0.000037166448
O -0.000149732118 -0.000267444730 0.000059048637
H 0.000024370430 -0.000005306667 0.000040796676
H 0.000017576882 -0.000061117703 0.000060935636
H 0.000040442983 0.000017174198 0.000033699239
H -0.000042785439 0.000178867332 0.000097831972
C -0.000114268137 0.000335407151 -0.000006449079
O 0.000048362130 -0.000103172856 0.000085227383
H -0.000016303299 0.000002480307 -0.000015984344
H -0.000036122666 -0.000055709034 0.000006306173
*** Time spent in gradient calculation: 1.65 sec ***
* Info * Energy : -268.1595559420 a.u.
* Info * Gradient : 2.149719e-04 a.u. (RMS)
* Info * 4.340613e-04 a.u. (Max)
* Info * Time : 4.05 sec
Optimization Step 44
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.039829775741 0.882920083275 0.059281138981
C -0.799668791507 0.136089589252 1.347656843393
O -0.060875641599 -0.816361470392 1.456111890890
H -1.009601874948 1.967416877448 0.273530653341
H -2.084265485108 0.666903073187 -0.240091955824
H -1.371178804983 0.508276700334 2.237950958047
H 0.114784730877 -1.353367189590 -0.593002952180
C -0.070302130956 0.492392300273 -1.059935921728
O -0.152433855176 -0.870337825484 -1.390339244500
H 0.957636936598 0.785617429643 -0.761167613471
H -0.312717170069 1.066217592198 -1.968885482704
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000039265615 -0.000081156849 -0.000308380469
C 0.000109502082 -0.000063305984 -0.000020476052
O -0.000068942723 -0.000102865179 0.000044668611
H 0.000002886795 -0.000009485437 0.000040791139
H -0.000034332606 -0.000008308137 0.000069816107
H -0.000002860630 0.000029677144 -0.000008340222
H -0.000005196279 0.000191182278 0.000107289112
C -0.000023660555 0.000233171919 0.000159126631
O -0.000064382321 -0.000076101215 -0.000042614028
H 0.000018535341 -0.000026229011 -0.000018774498
H 0.000002448665 -0.000101441027 -0.000007201183
*** Time spent in gradient calculation: 1.82 sec ***
* Info * Energy : -268.1595671267 a.u.
* Info * Gradient : 1.644938e-04 a.u. (RMS)
* Info * 3.212892e-04 a.u. (Max)
* Info * Time : 4.22 sec
Optimization Step 45
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.038373522580 0.883052695834 0.059533013342
C -0.802130898845 0.133947308955 1.347164448746
O -0.057290614743 -0.813519889798 1.457251031818
H -1.005583534938 1.967166080261 0.275743646726
H -2.082653539575 0.670725244871 -0.242697633002
H -1.382155785470 0.499191396284 2.234812765317
H 0.115215638720 -1.356185629731 -0.598821603981
C -0.068283554352 0.492264095875 -1.059163811332
O -0.153776540957 -0.869429653499 -1.393137619029
H 0.960070501456 0.781924152108 -0.758570097528
H -0.308370465800 1.069411982072 -1.966647155927
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000109829584 -0.000089373955 -0.000178733108
C 0.000014112906 0.000010383800 0.000070929280
O -0.000039438011 -0.000010986395 0.000014981565
H 0.000009395659 -0.000007953261 0.000039243771
H 0.000018731818 -0.000011347486 -0.000002694893
H -0.000018135929 -0.000028755769 -0.000005781982
H -0.000030300651 0.000086442168 0.000018453007
C -0.000067971443 0.000135668342 -0.000027939670
O -0.000020702034 -0.000079745201 0.000064356627
H 0.000019320498 -0.000007002676 -0.000015076613
H -0.000021809724 -0.000011124972 0.000033471972
*** Time spent in gradient calculation: 1.79 sec ***
* Info * Energy : -268.1595695231 a.u.
* Info * Gradient : 9.928773e-05 a.u. (RMS)
* Info * 2.280258e-04 a.u. (Max)
* Info * Time : 3.91 sec
Optimization Step 46
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.037696182366 0.883430028818 0.059503509282
C -0.801392776108 0.133096061021 1.346345911653
O -0.054231926194 -0.812618225760 1.456065520551
H -1.005162500802 1.967375360047 0.276653514905
H -2.081615604869 0.671189442060 -0.243973901752
H -1.382484086113 0.496495617233 2.234086312084
H 0.117359797665 -1.356223691489 -0.603015644117
C -0.065672418818 0.493698038586 -1.057965116924
O -0.152697925269 -0.867024319186 -1.395500237936
H 0.962324268632 0.780823235455 -0.754012640848
H -0.302305255329 1.073201951765 -1.964901344153
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000005293869 -0.000022933626 -0.000106113338
C -0.000046357370 -0.000042586922 -0.000065890240
O 0.000053405084 -0.000033066107 0.000044767852
H 0.000014615770 -0.000000117669 0.000005708261
H -0.000004740332 -0.000002286769 0.000018395284
H 0.000006884962 0.000013614250 0.000006531664
H 0.000010503744 0.000049012361 0.000049548806
C 0.000023329508 0.000074622007 0.000082310791
O -0.000083751960 -0.000016051765 -0.000013025552
H -0.000002522686 -0.000010634168 -0.000004586106
H 0.000006363055 -0.000022075895 -0.000009265759
*** Time spent in gradient calculation: 1.83 sec ***
* Info * Energy : -268.1595700463 a.u.
* Info * Gradient : 6.941759e-05 a.u. (RMS)
* Info * 1.135243e-04 a.u. (Max)
* Info * Time : 3.72 sec
Optimization Step 47
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.702919830311 1.017517647924 0.065452512242
C -0.386420146907 0.181923130783 1.280556154170
O 0.422537988989 -0.717909849753 1.297243645557
H -0.742371746143 2.079322775584 0.372086420806
H -1.736232027245 0.755948872632 -0.236253963857
H -0.968937879095 0.431482312267 2.205886458049
H 0.591068566625 -1.069332461227 -0.805705344486
C 0.267367710452 0.794213304642 -1.097867497936
O 0.269536121219 -0.535791841197 -1.549610034445
H 1.279617238235 1.128366455183 -0.788812813850
H -0.030935112984 1.429185263228 -1.947603417055
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000191197440 -0.000079860193 -0.000205223463
C 0.000085499025 -0.000081796101 -0.000016051318
O -0.000140620069 0.000013732831 0.000146252502
H 0.000013883694 -0.000005751700 -0.000043532131
H -0.000047962836 -0.000033657011 0.000089386275
H -0.000009044466 0.000003063425 -0.000011871650
H 0.000237973265 -0.000192782122 0.000492285425
C -0.000191214201 0.000151125244 0.000161605554
O -0.000235292137 0.000351304323 -0.000561139257
H 0.000055305535 -0.000057260470 -0.000035641543
H 0.000023531870 -0.000077365523 0.000000460565
*** Time spent in gradient calculation: 1.71 sec ***
* Info * Energy : -268.1595732015 a.u.
* Info * Gradient : 3.139048e-04 a.u. (RMS)
* Info * 7.026054e-04 a.u. (Max)
* Info * Time : 4.24 sec
Optimization Step 48
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.584266334110 1.018525559610 0.072375630712
C -0.212091416110 0.082365146856 1.194823875496
O 0.636990682071 -0.775927981750 1.113059318160
H -0.670308926243 2.041348506732 0.483997784257
H -1.607546323909 0.738226063458 -0.246048797503
H -0.791533566005 0.211399120777 2.146367042535
H 0.794141673541 -0.906163493268 -1.017250653694
C 0.379982058851 0.960179176407 -1.115874769403
O 0.438406959673 -0.315801959754 -1.700075090952
H 1.379545042291 1.309716591938 -0.783450286733
H 0.040730296868 1.662629036892 -1.894151114038
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000090284068 -0.000127368843 -0.000135599032
C 0.000155929810 -0.000075753308 0.000053248994
O -0.000194329231 0.000036462656 0.000103893033
H 0.000023699434 0.000010377513 -0.000047992780
H -0.000004664026 -0.000026292549 0.000039027635
H -0.000016070753 0.000014333013 -0.000018898672
H 0.000240500035 -0.000265339520 0.000386146351
C -0.000181654091 0.000137615912 0.000100621919
O -0.000211011433 0.000394920355 -0.000422170492
H 0.000059585547 -0.000055631282 -0.000059660295
H 0.000018020128 -0.000050629553 0.000013463567
*** Time spent in gradient calculation: 1.78 sec ***
* Info * Energy : -268.1595715234 a.u.
* Info * Gradient : 2.800927e-04 a.u. (RMS)
* Info * 6.153989e-04 a.u. (Max)
* Info * Time : 4.39 sec
Optimization Step 49
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.703154641314 1.017752478322 0.065404077427
C -0.385754395381 0.182001556427 1.280072179769
O 0.424360040557 -0.716874742153 1.295679523539
H -0.742974817989 2.079454455352 0.372424856758
H -1.736113454976 0.756027106608 -0.237109487140
H -0.967980131208 0.430492374894 2.205863623664
H 0.590409413608 -1.069523718448 -0.806188568971
C 0.267728401368 0.794490595557 -1.097757397252
O 0.269681556521 -0.535509345902 -1.549469637560
H 1.279755216668 1.128724457518 -0.788199255790
H -0.030314828671 1.429702991235 -1.947442610776
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000074361250 -0.000086267389 -0.000098496446
C 0.000045840400 -0.000022242697 0.000003808402
O -0.000068990975 -0.000001433217 0.000045352526
H 0.000003375518 -0.000000722414 -0.000015741994
H -0.000009225022 -0.000008385380 0.000041893208
H -0.000013416957 0.000015384153 -0.000009528016
H 0.000059710027 -0.000029246384 0.000114489658
C -0.000080752911 0.000107641246 0.000073379995
O -0.000066935440 0.000076908339 -0.000114979756
H 0.000025839061 -0.000024011496 -0.000029392761
H 0.000013034449 -0.000036182546 0.000004559423
*** Time spent in gradient calculation: 1.70 sec ***
* Info * Energy : -268.1595743794 a.u.
* Info * Gradient : 9.677740e-05 a.u. (RMS)
* Info * 1.536736e-04 a.u. (Max)
* Info * Time : 4.21 sec
Optimization Step 50
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.704332710582 1.017694285812 0.065817633617
C -0.386325351816 0.182654474063 1.280753033539
O 0.425198686122 -0.715007368605 1.296386027755
H -0.744265798989 2.079550923427 0.372328706825
H -1.737067077549 0.755766800665 -0.237178992191
H -0.968607393460 0.430471839379 2.206670662922
H 0.588976279169 -1.070855653088 -0.804701020701
C 0.267128834853 0.793305754891 -1.096976376072
O 0.268802262158 -0.536900692176 -1.547994470855
H 1.278939919015 1.127540741333 -0.786817877082
H -0.030410945139 1.428297267560 -1.947044458812
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000004339597 -0.000014779707 0.000009805524
C -0.000009991255 0.000022303007 -0.000014430183
O -0.000006391689 -0.000006005594 -0.000005343728
H -0.000010253334 0.000001405246 0.000008110490
H 0.000007048580 0.000003377831 -0.000000997196
H 0.000002210425 0.000006703060 0.000003729583
H -0.000025597671 0.000026871408 -0.000046874295
C 0.000006590348 0.000005734574 -0.000001902733
O 0.000022085902 -0.000048951005 0.000058707959
H -0.000001639320 0.000000753709 0.000000301577
H -0.000005869884 -0.000005149014 0.000002922569
*** Time spent in gradient calculation: 1.72 sec ***
* Info * Energy : -268.1595745466 a.u.
* Info * Gradient : 3.247516e-05 a.u. (RMS)
* Info * 7.956515e-05 a.u. (Max)
* Info * Time : 3.77 sec
Optimization Step 51
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -0.704402687975 1.017040511510 0.066502408016
C -0.384797442013 0.181268064083 1.280540252444
O 0.428503137403 -0.714784450343 1.295198649371
H -0.745207181367 2.078590324545 0.373958419226
H -1.737055721503 0.754323108720 -0.236137764760
H -0.967609419411 0.426878956474 2.206709740009
H 0.591221427125 -1.069301779183 -0.807064864911
C 0.266717734986 0.794793942593 -1.096994524626
O 0.269582422305 -0.534898930151 -1.549514500181
H 1.278326904887 1.129680739111 -0.786894376645
H -0.031814933923 1.430422204922 -1.946243880959
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000009216581 0.000008333886 0.000016624278
C -0.000004532113 0.000012371662 -0.000004185858
O -0.000011695441 0.000006391316 0.000003802908
H -0.000004078305 -0.000000321146 0.000001173966
H -0.000003663593 -0.000002211554 -0.000001565703
H 0.000002120676 -0.000000982448 0.000002627471
H 0.000005885139 -0.000016615347 0.000020929024
C -0.000001174310 -0.000015521723 -0.000002763188
O -0.000003707548 0.000005094292 -0.000026822743
H -0.000001181553 -0.000001849056 0.000005134869
H -0.000004074523 -0.000003195177 0.000000326615
*** Time spent in gradient calculation: 1.72 sec ***
* Info * Energy : -268.1595745014 a.u.
* Info * Gradient : 1.561220e-05 a.u. (RMS)
* Info * 2.755281e-05 a.u. (Max)
* Info * Time : 3.78 sec
Optimization Step 52
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.314498175821 0.950271102198 0.316054977108
C -0.549239033314 0.302473115508 1.267474871090
O -0.282553893430 -0.965848161574 1.204787229275
H -1.443406337737 2.032565963412 0.391787830066
H -2.093624362820 0.395271744304 -0.207500775360
H -0.007108988498 0.868479090460 2.043339001841
H -0.180461996477 -1.189169705359 0.200441057269
C -0.013069365773 0.397677603954 -1.237689389541
O 0.028844253699 -0.855745917912 -1.164907068594
H 0.865775023676 1.018375485836 -0.953429232525
H -0.679170610555 0.896410684667 -1.970952701374
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003141875984 -0.001400856074 -0.006813400388
C 0.002071015968 -0.003558414226 0.000662661175
O 0.000020094519 -0.000398683904 -0.015628274025
H 0.000639939383 -0.000146601851 -0.000330972644
H 0.000122411962 -0.000070869846 0.000230780081
H 0.000053046486 -0.000128862206 -0.000005024530
H 0.000462116279 0.004496676050 0.009064185946
C -0.005747200901 0.004941580935 0.006183620194
O -0.001134010423 -0.003468012188 0.006954776162
H -0.000012996407 -0.000011507414 0.000021414700
H 0.000403109383 -0.000298139684 -0.000333024517
*** Time spent in gradient calculation: 1.78 sec ***
* Info * Energy : -268.1020360783 a.u.
* Info * Gradient : 7.267657e-03 a.u. (RMS)
* Info * 1.563337e-02 a.u. (Max)
* Info * Time : 5.01 sec
Optimization Step 53
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.314501405349 0.950272539584 0.316061984971
C -0.549241163551 0.302476771276 1.267474190039
O -0.282553910801 -0.965847857572 1.204799297951
H -1.443414157996 2.032567712259 0.391791892752
H -2.093625840965 0.395272565671 -0.207503598842
H -0.007109619301 0.868480620115 2.043339071719
H -0.180467639660 -1.189224867535 0.200329968024
C -0.013063449626 0.397672516863 -1.237695748737
O 0.028845131957 -0.855743243087 -1.164912438026
H 0.865775207214 1.018375577495 -0.953429485033
H -0.679175525533 0.896414291102 -1.970948612207
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003146181042 -0.001403353951 -0.006801006695
C 0.002061176051 -0.003536661988 0.000659336855
O 0.000020264937 -0.000392441438 -0.015566909712
H 0.000639762894 -0.000146261266 -0.000331023777
H 0.000121230969 -0.000071875236 0.000228627139
H 0.000052487135 -0.000130305717 -0.000004647485
H 0.000463723367 0.004470945718 0.009007276511
C -0.005739890558 0.004917433150 0.006178622141
O -0.001132355341 -0.003445419116 0.006947510985
H -0.000013470815 -0.000010188336 0.000022121271
H 0.000400273232 -0.000295536262 -0.000333011116
*** Time spent in gradient calculation: 1.67 sec ***
* Info * Energy : -268.1020392466 a.u.
* Info * Gradient : 7.241588e-03 a.u. (RMS)
* Info * 1.557187e-02 a.u. (Max)
* Info * Time : 3.48 sec
Optimization Step 54
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.314506003401 0.950274586794 0.316071930567
C -0.549244177997 0.302481937173 1.267473226627
O -0.282553936002 -0.965847432335 1.204816390100
H -1.443425273615 2.032570192795 0.391797669889
H -2.093627922185 0.395273750936 -0.207507575400
H -0.007110506378 0.868482820182 2.043339164615
H -0.180475690900 -1.189302847978 0.200173013551
C -0.013055048711 0.397665319312 -1.237704782906
O 0.028846378813 -0.855739464889 -1.164920064273
H 0.865775476401 1.018375684910 -0.953429856135
H -0.679182464247 0.896419373348 -1.970942798248
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003152283678 -0.001406908591 -0.006783459270
C 0.002047317257 -0.003506045765 0.000654639868
O 0.000020499036 -0.000383528350 -0.015480351776
H 0.000639502164 -0.000145751602 -0.000331097033
H 0.000119556751 -0.000073294709 0.000225582713
H 0.000051675474 -0.000132352192 -0.000004139132
H 0.000465961231 0.004434612160 0.008927042101
C -0.005729538587 0.004883286145 0.006171554037
O -0.001130010802 -0.003413460687 0.006937214483
H -0.000014157605 -0.000008324224 0.000023125324
H 0.000396267105 -0.000291860221 -0.000332998654
*** Time spent in gradient calculation: 1.78 sec ***
* Info * Energy : -268.1020436986 a.u.
* Info * Gradient : 7.204847e-03 a.u. (RMS)
* Info * 1.548512e-02 a.u. (Max)
* Info * Time : 3.62 sec
Optimization Step 55
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.314512568319 0.950277511213 0.316086066242
C -0.549248444795 0.302489234586 1.267471863655
O -0.282553972902 -0.965846840600 1.204840611338
H -1.443441106469 2.032573714797 0.391805903911
H -2.093630846674 0.395275475019 -0.207513165644
H -0.007111750198 0.868486006166 2.043339284603
H -0.180487218907 -1.189413065447 0.199951348664
C -0.013043099019 0.397655133970 -1.237717641705
O 0.028848151837 -0.855734131119 -1.164930915406
H 0.865775876971 1.018375791854 -0.953430409428
H -0.679192252395 0.896426524174 -1.970934513302
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003160890964 -0.001411925627 -0.006758685924
C 0.002027733348 -0.003462841686 0.000647969033
O 0.000020861614 -0.000370979732 -0.015358263441
H 0.000639127242 -0.000145035905 -0.000331205708
H 0.000117209886 -0.000075283587 0.000221298581
H 0.000050538536 -0.000135224574 -0.000003403266
H 0.000469081599 0.004383325238 0.008813920626
C -0.005714913926 0.004835087038 0.006161546791
O -0.001126693964 -0.003368304932 0.006922651817
H -0.000015148322 -0.000005703443 0.000024537301
H 0.000390630500 -0.000286689952 -0.000332949338
*** Time spent in gradient calculation: 1.83 sec ***
* Info * Energy : -268.1020499371 a.u.
* Info * Gradient : 7.153108e-03 a.u. (RMS)
* Info * 1.536276e-02 a.u. (Max)
* Info * Time : 3.74 sec
Optimization Step 56
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.314522070408 0.950281747034 0.316106302620
C -0.549254492731 0.302499524143 1.267469935362
O -0.282554035172 -0.965846040512 1.204875041320
H -1.443463888416 2.032578743608 0.391817768049
H -2.093634921203 0.395278077115 -0.207520953712
H -0.007113471139 0.868490770091 2.043339410296
H -0.180504010669 -1.189568697242 0.199638933121
C -0.013025957886 0.397640706893 -1.237736114925
O 0.028850692437 -0.855726621998 -1.164946491807
H 0.865776515672 1.018375788098 -0.953431290799
H -0.679206010577 0.896436511204 -1.970922581095
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003173035414 -0.001419039763 -0.006723738021
C 0.002000164547 -0.003402225524 0.000638468750
O 0.000021393142 -0.000353162685 -0.015186459643
H 0.000638563852 -0.000143990919 -0.000331374189
H 0.000113944756 -0.000078041030 0.000215303747
H 0.000048926850 -0.000139238301 -0.000002355343
H 0.000473371630 0.004311081951 0.008654874665
C -0.005694253594 0.004767136044 0.006147377279
O -0.001122004097 -0.003304533845 0.006902036356
H -0.000016616273 -0.000002035819 0.000026507099
H 0.000382737923 -0.000279456656 -0.000332799865
*** Time spent in gradient calculation: 1.87 sec ***
* Info * Energy : -268.1020586456 a.u.
* Info * Gradient : 7.080467e-03 a.u. (RMS)
* Info * 1.519058e-02 a.u. (Max)
* Info * Time : 4.21 sec
Optimization Step 57
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.314535825735 0.950287885482 0.316135277804
C -0.549263064403 0.302514032946 1.267467207593
O -0.282554133107 -0.965844960829 1.204923982875
H -1.443496675647 2.032585922837 0.391834871113
H -2.093640593796 0.395281994749 -0.207531799381
H -0.007115849161 0.868497882797 2.043339525273
H -0.180528464887 -1.189788436552 0.199198674471
C -0.013001366491 0.397620273214 -1.237762658518
O 0.028854334011 -0.855716053421 -1.164968854645
H 0.865777529748 1.018375556865 -0.953432680414
H -0.679225345753 0.896450453320 -1.970905398422
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003190039471 -0.001428987536 -0.006674605365
C 0.001961192120 -0.003316835873 0.000624883627
O 0.000022309512 -0.000328379381 -0.014944764831
H 0.000637748966 -0.000142593294 -0.000331641817
H 0.000109447364 -0.000081839850 0.000206944207
H 0.000046743333 -0.000144775855 -0.000000738236
H 0.000479273810 0.004209351190 0.008431319551
C -0.005665150151 0.004671652702 0.006127275197
O -0.001115369609 -0.003214668834 0.006872911187
H -0.000018785868 0.000003058778 0.000029252319
H 0.000371740968 -0.000269391269 -0.000332405007
*** Time spent in gradient calculation: 1.77 sec ***
* Info * Energy : -268.1020707343 a.u.
* Info * Gradient : 6.978654e-03 a.u. (RMS)
* Info * 1.494839e-02 a.u. (Max)
* Info * Time : 4.04 sec
Optimization Step 58
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.314555936915 0.950296873591 0.316176995237
C -0.549275222308 0.302534459359 1.267463350695
O -0.282554292928 -0.965843538956 1.204993711062
H -1.443544223019 2.032596227079 0.391859736762
H -2.093648440714 0.395288019926 -0.207546795651
H -0.007119108266 0.868508708276 2.043339537934
H -0.180564505580 -1.190098418409 0.198579349731
C -0.012965862656 0.397591311749 -1.237801063747
O 0.028859584653 -0.855701214799 -1.165001173752
H 0.865779206355 1.018374768158 -0.953434940467
H -0.679252448008 0.896469805182 -1.970880482070
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003213848677 -0.001442935985 -0.006605586321
C 0.001906268600 -0.003197090321 0.000605417531
O 0.000023845868 -0.000293778071 -0.014605726244
H 0.000636533056 -0.000140669303 -0.000332073504
H 0.000103292393 -0.000087027181 0.000195340028
H 0.000043759853 -0.000152385863 0.000001710375
H 0.000487248704 0.004066409667 0.008118151156
C -0.005624160211 0.004537632715 0.006098758696
O -0.001105984650 -0.003088092485 0.006831734317
H -0.000022042574 0.000010107598 0.000033054334
H 0.000356477635 -0.000255447461 -0.000331535838
*** Time spent in gradient calculation: 1.63 sec ***
* Info * Energy : -268.1020873812 a.u.
* Info * Gradient : 6.836579e-03 a.u. (RMS)
* Info * 1.460870e-02 a.u. (Max)
* Info * Time : 3.58 sec
Optimization Step 59
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.314585766767 0.950310233615 0.316237548541
C -0.549292485604 0.302563149100 1.267457904090
O -0.282554564773 -0.965841749733 1.205093390239
H -1.443613937781 2.032611120343 0.391896342894
H -2.093659188847 0.395297542920 -0.207567297246
H -0.007123506871 0.868525613147 2.043339212567
H -0.180618502832 -1.190535041272 0.197710575881
C -0.012914126021 0.397550227301 -1.237857198672
O 0.028867221224 -0.855680466702 -1.165048336292
H 0.865782126696 1.018372691002 -0.953438752207
H -0.679290284144 0.896496423888 -1.970843997807
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003247023413 -0.001462391574 -0.006508972166
C 0.001829030290 -0.003029904340 0.000577387682
O 0.000026486980 -0.000245732939 -0.014131878088
H 0.000634681428 -0.000138059229 -0.000332794920
H 0.000095005424 -0.000093985080 0.000179379838
H 0.000039749780 -0.000162691919 0.000005495597
H 0.000497781257 0.003866203676 0.007681325948
C -0.005566555144 0.004350268034 0.006058272692
O -0.001092712274 -0.002910231472 0.006773545360
H -0.000027026981 0.000019755676 0.000038266896
H 0.000335483171 -0.000236330346 -0.000329675873
*** Time spent in gradient calculation: 1.91 sec ***
* Info * Energy : -268.1021100384 a.u.
* Info * Gradient : 6.639589e-03 a.u. (RMS)
* Info * 1.413404e-02 a.u. (Max)
* Info * Time : 3.84 sec
Optimization Step 60
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.314630948667 0.950330530634 0.316326527529
C -0.549317030563 0.302603284784 1.267450238086
O -0.282555051220 -0.965839695902 1.205236611153
H -1.443717823283 2.032632893438 0.391951255437
H -2.093673703408 0.395313107759 -0.207594815588
H -0.007129308488 0.868552889111 2.043337917446
H -0.180701269922 -1.191148355991 0.196497703158
C -0.012837679572 0.397491871841 -1.237940497131
O 0.028878472614 -0.855651664024 -1.165118160575
H 0.865787558805 1.018367718782 -0.953445451701
H -0.679342790010 0.896532525745 -1.970789879666
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003292808013 -0.001489227839 -0.006374562136
C 0.001720804788 -0.002798137291 0.000536794308
O 0.000031157284 -0.000179733368 -0.013473290784
H 0.000631785269 -0.000134551554 -0.000334047319
H 0.000084126090 -0.000103064661 0.000157730115
H 0.000034547381 -0.000176258073 0.000011550707
H 0.000511194940 0.003587241306 0.007075955902
C -0.005485847026 0.004089966449 0.006000684354
O -0.001073980010 -0.002661244117 0.006691436591
H -0.000034862634 0.000032718463 0.000045310058
H 0.000307025858 -0.000210578748 -0.000325728268
*** Time spent in gradient calculation: 1.72 sec ***
* Info * Energy : -268.1021403493 a.u.
* Info * Gradient : 6.369175e-03 a.u. (RMS)
* Info * 1.347453e-02 a.u. (Max)
* Info * Time : 4.27 sec
Optimization Step 61
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.314701509952 0.950362460247 0.316459803601
C -0.549351921299 0.302659106439 1.267439513670
O -0.282555699067 -0.965837700305 1.205443777805
H -1.443876547592 2.032665365837 0.392036261207
H -2.093692779403 0.395339672924 -0.207630698494
H -0.007136785171 0.868598746520 2.043333992494
H -0.180832452333 -1.192005698172 0.194819361787
C -0.012722349627 0.397408872235 -1.238066961162
O 0.028895421734 -0.855612189400 -1.165223813898
H 0.865798533329 1.018356223487 -0.953457792420
H -0.679414996611 0.896580393358 -1.970708290162
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003355384919 -0.001525656655 -0.006189466981
C 0.001570462603 -0.002481245511 0.000477642141
O 0.000039390640 -0.000089582405 -0.012566237519
H 0.000627073643 -0.000130130427 -0.000336325760
H 0.000070490007 -0.000114280195 0.000129141749
H 0.000027912979 -0.000193513073 0.000021229070
H 0.000527141708 0.003201966050 0.006245848662
C -0.005373699555 0.003732903037 0.005919082812
O -0.001047534896 -0.002315856077 0.006575860107
H -0.000047415881 0.000049660063 0.000054555419
H 0.000269311436 -0.000176849210 -0.000317582022
*** Time spent in gradient calculation: 1.84 sec ***
* Info * Energy : -268.1021798678 a.u.
* Info * Gradient : 6.004286e-03 a.u. (RMS)
* Info * 1.256662e-02 a.u. (Max)
* Info * Time : 4.06 sec
Optimization Step 62
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.314816875969 0.950414964187 0.316665433983
C -0.549401589695 0.302735645347 1.267424884853
O -0.282557037629 -0.965837291769 1.205747209273
H -1.444127732511 2.032715335455 0.392173642492
H -2.093717295142 0.395386989209 -0.207674745743
H -0.007145749973 0.868679740829 2.043322514837
H -0.181049166392 -1.193189487445 0.192539298988
C -0.012542507737 0.397290534174 -1.238265803101
O 0.028921702907 -0.855559637545 -1.165389164092
H 0.865822972665 1.018330072936 -0.953481714597
H -0.679512899533 0.896641493519 -1.970582952101
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003438866379 -0.001573043269 -0.005940049410
C 0.001365254976 -0.002059400795 0.000391016342
O 0.000053905296 0.000031164619 -0.011336431696
H 0.000619030450 -0.000124872758 -0.000340674941
H 0.000054689222 -0.000126817650 0.000093089920
H 0.000020005350 -0.000213615803 0.000037062471
H 0.000543661274 0.002679256284 0.005128213055
C -0.005219927833 0.003254603875 0.005804218584
O -0.001010524985 -0.001845098281 0.006414391314
H -0.000068043687 0.000070643337 0.000066132079
H 0.000221306939 -0.000135077559 -0.000300883147
*** Time spent in gradient calculation: 1.64 sec ***
* Info * Energy : -268.1022294280 a.u.
* Info * Gradient : 5.526747e-03 a.u. (RMS)
* Info * 1.133660e-02 a.u. (Max)
* Info * Time : 3.55 sec
Optimization Step 63
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.315018230587 0.950507148709 0.316997702057
C -0.549471940323 0.302837797441 1.267406300182
O -0.282559882816 -0.965842732302 1.206198543123
H -1.444546069177 2.032796532340 0.392411858664
H -2.093748000080 0.395475336801 -0.207722616680
H -0.007155239157 0.868830661715 2.043288620249
H -0.181427616863 -1.194775690800 0.189579717663
C -0.012247902281 0.397121539668 -1.238595237503
O 0.028964417401 -0.855494334016 -1.165661256658
H 0.865883975452 1.018270230181 -0.953530747119
H -0.679642641180 0.896714964460 -1.970388186790
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003545370805 -0.001628180174 -0.005620712102
C 0.001097914718 -0.001532244284 0.000265519020
O 0.000078428035 0.000188434173 -0.009723367502
H 0.000604475495 -0.000119456287 -0.000349358025
H 0.000039017677 -0.000137799734 0.000051929394
H 0.000011195953 -0.000232681632 0.000062433670
H 0.000555093116 0.001998337749 0.003679140763
C -0.005016309774 0.002648962626 0.005647488212
O -0.000959911721 -0.001231974774 0.006194002820
H -0.000101967270 0.000093974897 0.000079302768
H 0.000164605471 -0.000089302422 -0.000267741978
*** Time spent in gradient calculation: 1.66 sec ***
* Info * Energy : -268.1022880746 a.u.
* Info * Gradient : 4.940137e-03 a.u. (RMS)
* Info * 9.725509e-03 a.u. (Max)
* Info * Time : 3.67 sec
Optimization Step 64
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.315395643013 0.950679417446 0.317565099641
C -0.549569527253 0.302966940252 1.267388334876
O -0.282567299001 -0.965866850653 1.206874340815
H -1.445277371188 2.032938209118 0.392861863614
H -2.093787539369 0.395643181908 -0.207762012397
H -0.007160762928 0.869119113920 2.043188016596
H -0.182118417816 -1.196724402813 0.186225223952
C -0.011737280510 0.396881119329 -1.239174488776
O 0.029037396804 -0.855427676134 -1.166134743687
H 0.866050473740 1.018134677522 -0.953634717101
H -0.679807137352 0.896799610343 -1.970095845719
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003669244083 -0.001671413928 -0.005267312230
C 0.000795115473 -0.000980506298 0.000096266264
O 0.000114053180 0.000381360951 -0.007781716227
H 0.000577166907 -0.000115254842 -0.000366772122
H 0.000027131988 -0.000141298354 0.000015473810
H 0.000001023662 -0.000240342052 0.000098036189
H 0.000550002652 0.001205489765 0.001970084306
C -0.004773062673 0.001992368722 0.005458270164
O -0.000896139916 -0.000533358209 0.005916278593
H -0.000153208823 0.000114574922 0.000091312579
H 0.000106368266 -0.000053262929 -0.000209110124
*** Time spent in gradient calculation: 1.63 sec ***
* Info * Energy : -268.1023525289 a.u.
* Info * Gradient : 4.319735e-03 a.u. (RMS)
* Info * 7.791890e-03 a.u. (Max)
* Info * Time : 3.89 sec
Optimization Step 65
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.316097599869 0.950990945670 0.318535069299
C -0.549698812314 0.303125751013 1.267387882592
O -0.282584704405 -0.965937666064 1.207830964242
H -1.446508647707 2.033187507832 0.393728909457
H -2.093837954671 0.395934918348 -0.207788952688
H -0.007143078771 0.869617215550 2.042932481888
H -0.183317685462 -1.198674965552 0.183736381659
C -0.010858855817 0.396543558081 -1.240188004274
O 0.029160319255 -0.855389060521 -1.166949235014
H 0.866472620319 1.017858042826 -0.953837922839
H -0.679994458868 0.896921878702 -1.969723455792
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003794075061 -0.001656246997 -0.005016958350
C 0.000567510999 -0.000646481413 -0.000083135767
O 0.000147160425 0.000587125419 -0.005896237755
H 0.000530731517 -0.000112349314 -0.000396881751
H 0.000016727457 -0.000133298975 0.000002377473
H -0.000014673563 -0.000222611237 0.000130244718
H 0.000519989288 0.000517280078 0.000367053396
C -0.004557815741 0.001535516105 0.005310990795
O -0.000833373112 0.000011651632 0.005641753189
H -0.000210026362 0.000126707476 0.000097028498
H 0.000057456665 -0.000048580911 -0.000134485143
*** Time spent in gradient calculation: 1.73 sec ***
* Info * Energy : -268.1024220334 a.u.
* Info * Gradient : 3.853590e-03 a.u. (RMS)
* Info * 7.165062e-03 a.u. (Max)
* Info * Time : 3.89 sec
Optimization Step 66
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.317252929219 0.951468856709 0.320064698673
C -0.549877390352 0.303367131521 1.267426887381
O -0.282615795240 -0.966089688595 1.209048317296
H -1.448287523819 2.033577086068 0.395233929541
H -2.093865715048 0.396357629758 -0.207870760905
H -0.007049089590 0.870293266572 2.042465116265
H -0.185108465207 -1.200311328501 0.183808950555
C -0.009492174821 0.396048805469 -1.241795043906
O 0.029347461081 -0.855382969283 -1.168206703351
H 0.867295542078 1.017386655637 -0.954154958572
H -0.680171507773 0.897187889513 -1.969341716808
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003926464383 -0.001562422577 -0.005031792795
C 0.000528806950 -0.000724657869 -0.000207080863
O 0.000152558772 0.000765316216 -0.004583915075
H 0.000470919356 -0.000108211771 -0.000434955425
H -0.000007330968 -0.000121808113 0.000021271276
H -0.000039900754 -0.000182922573 0.000135783392
H 0.000483754098 0.000175366227 -0.000699365131
C -0.004476614526 0.001503779589 0.005331427551
O -0.000796458806 0.000154335982 0.005481779407
H -0.000242797568 0.000135590374 0.000092734298
H 0.000018810783 -0.000074850995 -0.000084723223
*** Time spent in gradient calculation: 1.67 sec ***
* Info * Energy : -268.1025094217 a.u.
* Info * Gradient : 3.673297e-03 a.u. (RMS)
* Info * 7.122187e-03 a.u. (Max)
* Info * Time : 3.75 sec
Optimization Step 67
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.318930714309 0.952109541258 0.322285467837
C -0.550147723036 0.303789761047 1.267502943713
O -0.282656760156 -0.966336383360 1.210578128159
H -1.450593923298 2.034113959658 0.397485058063
H -2.093785885131 0.396926865336 -0.208118730823
H -0.006837315392 0.871091620080 2.041831330554
H -0.187567257790 -1.202160412146 0.186333682951
C -0.007549836023 0.395288550728 -1.244110716275
O 0.029607947264 -0.855355165530 -1.169971148899
H 0.868507496951 1.016685191176 -0.954552912437
H -0.680334262407 0.897689400775 -1.968882237274
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.004110454705 -0.001444364900 -0.005274598018
C 0.000605963436 -0.001042494046 -0.000292849652
O 0.000134946036 0.000918611708 -0.003679494728
H 0.000408067679 -0.000104119783 -0.000476380123
H -0.000045610092 -0.000109616561 0.000062446377
H -0.000067495006 -0.000134595644 0.000124643864
H 0.000462344535 0.000053184627 -0.001433898576
C -0.004528394421 0.001734188037 0.005524910792
O -0.000787719565 0.000056562226 0.005445505742
H -0.000260896648 0.000147664028 0.000078737842
H -0.000012726648 -0.000114011964 -0.000059614465
*** Time spent in gradient calculation: 1.76 sec ***
* Info * Energy : -268.1026340801 a.u.
* Info * Gradient : 3.691581e-03 a.u. (RMS)
* Info * 7.351082e-03 a.u. (Max)
* Info * Time : 4.06 sec
Optimization Step 68
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.321261660942 0.952826801379 0.325454702085
C -0.550613613922 0.304700770825 1.267650127457
O -0.282684489924 -0.966734063804 1.212123240953
H -1.452919134955 2.034768944701 0.400752386190
H -2.093164459392 0.397522935806 -0.208995204808
H -0.006302894289 0.871607256490 2.041214895301
H -0.190710489933 -1.204516694647 0.194894191224
C -0.004948298410 0.393993349667 -1.247325180525
O 0.029949212177 -0.855170537412 -1.172256077467
H 0.870141858780 1.015608115012 -0.954848130115
H -0.680305386431 0.898769259726 -1.968375781161
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.004417325282 -0.001425555130 -0.006008160736
C 0.000852479318 -0.001573404374 -0.000224808274
O 0.000075737077 0.000843506420 -0.004611255903
H 0.000405733842 -0.000109672659 -0.000490441577
H -0.000103726284 -0.000118590962 0.000124905752
H -0.000066058673 -0.000116302538 0.000089991138
H 0.000528816826 0.000514454695 -0.000772750455
C -0.005007379958 0.002351234087 0.006135505584
O -0.000864366695 -0.000430169219 0.005843600196
H -0.000225938426 0.000169811046 0.000047468227
H 0.000007267700 -0.000141671070 -0.000120136706
*** Time spent in gradient calculation: 1.66 sec ***
* Info * Energy : -268.1028228174 a.u.
* Info * Gradient : 4.133510e-03 a.u. (RMS)
* Info * 8.261149e-03 a.u. (Max)
* Info * Time : 3.79 sec
Optimization Step 69
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.324667095661 0.953929210338 0.330090076971
C -0.551245480635 0.305860228306 1.267828821192
O -0.282727417950 -0.967201445830 1.214753731786
H -1.456616250845 2.035761944886 0.405203964752
H -2.092212624762 0.398559907627 -0.210224449619
H -0.005766478506 0.872671236244 2.040382023481
H -0.195597324065 -1.208901253500 0.202076203578
C -0.001094303430 0.392210774795 -1.252047219680
O 0.030446854860 -0.854956269119 -1.175603015676
H 0.872312608548 1.013971051387 -0.955196003707
H -0.680411405348 0.900003599908 -1.967323223995
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.004782861925 -0.001421880380 -0.006555335154
C 0.000931460323 -0.001898891701 -0.000363486996
O 0.000050773660 0.000972576270 -0.003961673137
H 0.000373511883 -0.000121054420 -0.000547519895
H -0.000105177450 -0.000087836458 0.000194715501
H -0.000073714330 -0.000067060969 0.000093857188
H 0.000547421148 0.000417479053 -0.001455918853
C -0.005297557981 0.002667246944 0.006574768070
O -0.000901190223 -0.000453674019 0.006053453394
H -0.000294657437 0.000165433517 0.000031532839
H 0.000006959236 -0.000205854710 -0.000055706222
*** Time spent in gradient calculation: 1.74 sec ***
* Info * Energy : -268.1031138466 a.u.
* Info * Gradient : 4.353376e-03 a.u. (RMS)
* Info * 8.854711e-03 a.u. (Max)
* Info * Time : 4.22 sec
Optimization Step 70
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.329335015986 0.955142435573 0.336668764112
C -0.552311731200 0.308104512570 1.268155669723
O -0.282708617311 -0.967920124271 1.217419903222
H -1.460207827467 2.037013623152 0.411580576863
H -2.090181079305 0.399597312878 -0.212947893851
H -0.004743008725 0.872960461600 2.039749419476
H -0.202091275234 -1.215554611106 0.219279954634
C 0.004045159178 0.389245626326 -1.258532105682
O 0.031113380925 -0.854359915472 -1.179913612568
H 0.875400905919 1.011613911258 -0.955296067501
H -0.680215794852 0.902554130082 -1.966305172504
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005547015202 -0.001675988654 -0.007588729002
C 0.001098529290 -0.001941228469 -0.000258757550
O 0.000036188390 0.000389635506 -0.006996237270
H 0.000475788353 -0.000127936604 -0.000546275405
H -0.000243947073 -0.000192834278 0.000167548263
H -0.000059998234 -0.000155521202 0.000011413258
H 0.000699097057 0.001212205998 0.000731491682
C -0.006233703250 0.002874367422 0.007729341428
O -0.001065372138 -0.000569178111 0.006947532959
H -0.000188817901 0.000262224005 0.000037331295
H -0.000043991903 -0.000107239596 -0.000242821551
*** Time spent in gradient calculation: 1.74 sec ***
* Info * Energy : -268.1035499903 a.u.
* Info * Gradient : 5.267964e-03 a.u. (RMS)
* Info * 1.033749e-02 a.u. (Max)
* Info * Time : 4.00 sec
Optimization Step 71
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.333803619119 0.956649939786 0.342641131271
C -0.553137489828 0.309100344630 1.268221390065
O -0.282740289275 -0.967556887002 1.223527878856
H -1.465545519295 2.038174202141 0.415769613682
H -2.086916755508 0.402026041741 -0.213805351943
H -0.004571431713 0.875668205952 2.040528335229
H -0.209896445832 -1.234417077396 0.183141078729
C 0.009042653380 0.387438816351 -1.264783203387
O 0.031821560368 -0.854160881181 -1.184069763069
H 0.876042206694 1.008025532530 -0.955786248718
H -0.679157328225 0.901765717986 -1.962356796489
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.004793513034 -0.002882687227 -0.005058228290
C -0.001805780246 0.002462530652 -0.002351302536
O 0.000690006964 0.003584898252 0.012534890757
H 0.000453126212 -0.000313157970 -0.000735889181
H 0.001168628148 0.000667440360 0.000686606467
H 0.000219620469 0.000097563760 0.000912265260
H 0.000087476157 -0.007084736763 -0.016650536623
C -0.004462129438 -0.001204937195 0.004559005630
O -0.000258908160 0.005634635723 0.004599708549
H -0.001511730850 -0.000300819233 -0.000045955298
H 0.000649873877 -0.000689105053 0.001562738533
*** Time spent in gradient calculation: 1.70 sec ***
* Info * Energy : -268.1031209253 a.u.
* Info * Gradient : 7.830446e-03 a.u. (RMS)
* Info * 1.809535e-02 a.u. (Max)
* Info * Time : 4.09 sec
Optimization Step 72
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.329981992984 0.955356018417 0.337543948196
C -0.552433239702 0.308275540793 1.268173883465
O -0.282711814143 -0.967896640948 1.218219470291
H -1.460949357906 2.037184975931 0.412240002956
H -2.089749008742 0.399912083675 -0.213105607536
H -0.004693465736 0.873294604164 2.039822121340
H -0.203155493377 -1.217939362723 0.215371477466
C 0.004773006743 0.388958361630 -1.259438717408
O 0.031211823726 -0.854326239366 -1.180520206931
H 0.875555689720 1.011140709186 -0.955361872725
H -0.680087564138 0.902524446889 -1.965810556626
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005472411915 -0.001821297513 -0.007312953464
C 0.000759743104 -0.001425412288 -0.000494609464
O 0.000061462998 0.000857116110 -0.004324556117
H 0.000474473994 -0.000149831053 -0.000569642432
H -0.000079297456 -0.000093263808 0.000227929716
H -0.000026463523 -0.000125748177 0.000116326162
H 0.000687782046 0.000158894407 -0.001716584034
C -0.006037352354 0.002398844990 0.007381707311
O -0.000981993963 0.000142068388 0.006695819158
H -0.000342737838 0.000200481337 0.000028416895
H 0.000033063221 -0.000171767962 -0.000033739890
*** Time spent in gradient calculation: 1.75 sec ***
* Info * Energy : -268.1036134890 a.u.
* Info * Gradient : 4.822805e-03 a.u. (RMS)
* Info * 9.833295e-03 a.u. (Max)
* Info * Time : 4.17 sec
Optimization Step 73
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.329429249197 0.955171431326 0.336797515385
C -0.552330186356 0.308135904329 1.268159736313
O -0.282709100480 -0.967923524871 1.217514504304
H -1.460306118780 2.037038904220 0.411688264210
H -2.090128704099 0.399637176203 -0.212979836993
H -0.004731423087 0.872994796887 2.039749581989
H -0.202235155942 -1.215820569449 0.219049198019
C 0.004151202990 0.389198061252 -1.258663651760
O 0.031127098317 -0.854353147761 -1.180002247600
H 0.875436447101 1.011557211935 -0.955304120288
H -0.680204567148 0.902570299802 -1.966251570822
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005544916447 -0.001691081703 -0.007571195507
C 0.001069463390 -0.001895105443 -0.000279202036
O 0.000037723918 0.000428027228 -0.006771658928
H 0.000475976267 -0.000129856134 -0.000548604296
H -0.000229424575 -0.000183766988 0.000173331949
H -0.000057140387 -0.000153431257 0.000020022253
H 0.000699750301 0.001121834305 0.000522128848
C -0.006220229571 0.002832143093 0.007705986677
O -0.001059553832 -0.000505771460 0.006930250840
H -0.000202614382 0.000257724928 0.000036499753
H -0.000038052342 -0.000112056486 -0.000225253940
*** Time spent in gradient calculation: 1.71 sec ***
* Info * Energy : -268.1035603446 a.u.
* Info * Gradient : 5.223772e-03 a.u. (RMS)
* Info * 1.030022e-02 a.u. (Max)
* Info * Time : 3.85 sec
Optimization Step 74
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.329569199403 0.955214274608 0.336988597684
C -0.552356810028 0.308183217635 1.268167051772
O -0.282709872517 -0.967932125614 1.217640827448
H -1.460448709844 2.037077449533 0.411853565085
H -2.090060985456 0.399690469479 -0.213032929453
H -0.004713922969 0.873039633614 2.039742227086
H -0.202445213647 -1.216146346720 0.218922430101
C 0.004308274259 0.389126859578 -1.258858035710
O 0.031147089396 -0.854344169588 -1.180133551154
H 0.875500027566 1.011479505925 -0.955315181008
H -0.680193236928 0.902603974295 -1.966186424972
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005547364642 -0.001706142736 -0.007558989646
C 0.001041893176 -0.001851894256 -0.000300863090
O 0.000039141882 0.000465917245 -0.006550402192
H 0.000476193856 -0.000131871380 -0.000551684639
H -0.000215420000 -0.000175033574 0.000178921580
H -0.000054465320 -0.000151151004 0.000028345185
H 0.000700496721 0.001032467757 0.000314446697
C -0.006209559560 0.002793105093 0.007688106891
O -0.001054664779 -0.000443235110 0.006916515319
H -0.000217161780 0.000253566271 0.000035846399
H -0.000032974812 -0.000116903697 -0.000207567596
*** Time spent in gradient calculation: 1.67 sec ***
* Info * Energy : -268.1035748770 a.u.
* Info * Gradient : 5.184582e-03 a.u. (RMS)
* Info * 1.026972e-02 a.u. (Max)
* Info * Time : 3.57 sec
Optimization Step 75
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.329769772405 0.955276242619 0.337261834226
C -0.552393851427 0.308249314806 1.268178434317
O -0.282711035324 -0.967945575480 1.217815166852
H -1.460653064086 2.037133659396 0.412091865768
H -2.089970442840 0.399762787767 -0.213111428533
H -0.004690082031 0.873102687319 2.039727824414
H -0.202746296560 -1.216570796243 0.218838863635
C 0.004532863854 0.389026367401 -1.259135754905
O 0.031175544317 -0.854333196616 -1.180321220213
H 0.875598064653 1.011370235334 -0.955330766735
H -0.680179284326 0.902654570340 -1.966101580114
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005553302329 -0.001722322879 -0.007547366531
C 0.001010967203 -0.001805276382 -0.000327994713
O 0.000040724530 0.000511849703 -0.006285762411
H 0.000476315793 -0.000134324795 -0.000556085672
H -0.000199698682 -0.000165464077 0.000184887945
H -0.000051471396 -0.000147886779 0.000038146382
H 0.000700924767 0.000927032493 0.000066248089
C -0.006198476214 0.002751203658 0.007669555251
O -0.001049275141 -0.000371142838 0.006902086204
H -0.000234950374 0.000248593716 0.000035362140
H -0.000027483585 -0.000123233923 -0.000186022916
*** Time spent in gradient calculation: 1.76 sec ***
* Info * Energy : -268.1035951581 a.u.
* Info * Gradient : 5.141866e-03 a.u. (RMS)
* Info * 1.023779e-02 a.u. (Max)
* Info * Time : 3.66 sec
Optimization Step 76
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.330056876544 0.955365622029 0.337651956315
C -0.552445247257 0.308341654092 1.268196213372
O -0.282712765900 -0.967966707318 1.218053736761
H -1.460945148605 2.037215586356 0.412435675399
H -2.089850388785 0.399860583989 -0.213227012524
H -0.004657669329 0.873190372879 2.039701239326
H -0.203176602127 -1.217111812026 0.218877729363
C 0.004853442909 0.388884623314 -1.259531859922
O 0.031215943988 -0.854320287827 -1.180588986414
H 0.875749519732 1.011217330844 -0.955352883444
H -0.680162458790 0.902731263511 -1.965993996802
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005565199945 -0.001737653769 -0.007540629186
C 0.000980267759 -0.001762271419 -0.000360384522
O 0.000042221135 0.000564016589 -0.005991139437
H 0.000476216041 -0.000137069624 -0.000562298459
H -0.000183542083 -0.000156002420 0.000190627379
H -0.000048534479 -0.000143364238 0.000048707011
H 0.000700709684 0.000812167366 -0.000210034579
C -0.006189555128 0.002713041040 0.007654550200
O -0.001044185423 -0.000295657879 0.006889988370
H -0.000255374596 0.000243205550 0.000035377209
H -0.000022502172 -0.000131143143 -0.000161406002
*** Time spent in gradient calculation: 1.84 sec ***
* Info * Energy : -268.1036235083 a.u.
* Info * Gradient : 5.100985e-03 a.u. (RMS)
* Info * 1.021094e-02 a.u. (Max)
* Info * Time : 3.81 sec
Optimization Step 77
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.330466708248 0.955493445165 0.338207051302
C -0.552516691449 0.308470995754 1.268223714527
O -0.282715129358 -0.967999575246 1.218377755043
H -1.461360168535 2.037334136753 0.412931777048
H -2.089691753098 0.399993476973 -0.213395603956
H -0.004613570401 0.873310719190 2.039654957663
H -0.203788106459 -1.217790241401 0.219164104967
C 0.005309122225 0.388684892107 -1.260095051494
O 0.031273445054 -0.854305193285 -1.180969610740
H 0.875981931482 1.011003741736 -0.955384832510
H -0.680141632939 0.902847825708 -1.965861225548
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005586171187 -0.001749851154 -0.007546048081
C 0.000955552511 -0.001733434279 -0.000397090331
O 0.000043181110 0.000618369190 -0.005692829116
H 0.000475795917 -0.000139793380 -0.000570861319
H -0.000168539071 -0.000147743205 0.000195589749
H -0.000046252415 -0.000137378459 0.000058680544
H 0.000699559762 0.000699969744 -0.000490293283
C -0.006187414815 0.002688568105 0.007649901698
O -0.001040651592 -0.000226624167 0.006885333105
H -0.000277086474 0.000238045098 0.000036371585
H -0.000019347572 -0.000140589253 -0.000135301957
*** Time spent in gradient calculation: 1.82 sec ***
* Info * Energy : -268.1036633005 a.u.
* Info * Gradient : 5.069800e-03 a.u. (RMS)
* Info * 1.019968e-02 a.u. (Max)
* Info * Time : 3.92 sec
Optimization Step 78
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.331049147346 0.955674736972 0.338994370658
C -0.552616593522 0.308653814740 1.268265690136
O -0.282718454128 -0.968049671830 1.218818355946
H -1.461946287577 2.037504104247 0.413644710055
H -2.089479073785 0.400176685432 -0.213637820254
H -0.004552216261 0.873473518716 2.039581266809
H -0.204651191698 -1.218652272975 0.219839875459
C 0.005954711502 0.388402285587 -1.260892901814
O 0.031354605466 -0.854286946365 -1.181508626232
H 0.876330769568 1.010706177720 -0.955431900958
H -0.680112978866 0.903023492605 -1.965697505344
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005619713287 -0.001758159245 -0.007571519464
C 0.000941342436 -0.001726197201 -0.000437939326
O 0.000043227774 0.000671150965 -0.005410991709
H 0.000475024955 -0.000142232750 -0.000582482859
H -0.000155266568 -0.000141076074 0.000199768827
H -0.000045091100 -0.000129895189 0.000066905107
H 0.000697571346 0.000599271442 -0.000757033056
C -0.006196577436 0.002684535018 0.007663143437
O -0.001040207364 -0.000170334912 0.006893514280
H -0.000299331733 0.000234077990 0.000038741551
H -0.000019380639 -0.000151302125 -0.000108907704
*** Time spent in gradient calculation: 1.71 sec ***
* Info * Energy : -268.1037195042 a.u.
* Info * Gradient : 5.054620e-03 a.u. (RMS)
* Info * 1.021411e-02 a.u. (Max)
* Info * Time : 3.81 sec
Optimization Step 79
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.331605653778 0.955841323092 0.339723241716
C -0.552706818383 0.308839665417 1.268359140320
O -0.282721899038 -0.968150403895 1.218945494696
H -1.462426475510 2.037682636511 0.414516159125
H -2.089468715941 0.400196353912 -0.213877601694
H -0.004460508364 0.873477287621 2.039405693321
H -0.205321047848 -1.218085851958 0.224343543622
C 0.006520974826 0.388136355494 -1.261617152846
O 0.031433283247 -0.854305422699 -1.181997119568
H 0.877032191685 1.010456066595 -0.955539189939
H -0.680010027655 0.903367983924 -1.965983068321
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005713125177 -0.001639940037 -0.007859007634
C 0.001202545615 -0.002155478949 -0.000305868643
O 0.000026332372 0.000338449092 -0.007226354635
H 0.000479100442 -0.000124866333 -0.000576289759
H -0.000263139632 -0.000218273206 0.000161490402
H -0.000067203909 -0.000142784870 -0.000007034973
H 0.000697148450 0.001342168663 0.000913479387
C -0.006443711090 0.003095498535 0.007960163814
O -0.001088889518 -0.000694480687 0.007110518162
H -0.000163159345 0.000288495483 0.000056805069
H -0.000071176629 -0.000119908619 -0.000240826185
*** Time spent in gradient calculation: 1.63 sec ***
* Info * Energy : -268.1037653501 a.u.
* Info * Gradient : 5.446994e-03 a.u. (RMS)
* Info * 1.069896e-02 a.u. (Max)
* Info * Time : 3.77 sec
Optimization Step 80
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.331146772223 0.955705222208 0.339125789277
C -0.552632851595 0.308683892276 1.268273814747
O -0.282719014027 -0.968058990634 1.218886147887
H -1.462043494480 2.037533189460 0.413767042890
H -2.089448241268 0.400203975995 -0.213679341680
H -0.004542119710 0.873498354465 2.039566340487
H -0.204793863254 -1.218763887747 0.220033248642
C 0.006062023280 0.388355583477 -1.261025757812
O 0.031368161687 -0.854285110018 -1.181598359613
H 0.876396654424 1.010657492995 -0.955440635027
H -0.680107807334 0.903055973230 -1.965679213238
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005626890963 -0.001756060203 -0.007580826495
C 0.000945354591 -0.001735590836 -0.000441296169
O 0.000042781152 0.000672571918 -0.005406556173
H 0.000474895084 -0.000142228643 -0.000584203015
H -0.000156083919 -0.000142076550 0.000199156064
H -0.000045529482 -0.000128963341 0.000066456649
H 0.000697119887 0.000600460202 -0.000761860191
C -0.006202471173 0.002693608099 0.007671329680
O -0.001041271712 -0.000174256271 0.006899211962
H -0.000299820593 0.000234617220 0.000039555588
H -0.000020834049 -0.000152218801 -0.000107929653
*** Time spent in gradient calculation: 1.53 sec ***
* Info * Energy : -268.1037288967 a.u.
* Info * Gradient : 5.059578e-03 a.u. (RMS)
* Info * 1.022621e-02 a.u. (Max)
* Info * Time : 3.61 sec
Optimization Step 81
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.331284979974 0.955748331230 0.339312152490
C -0.552656034097 0.308726563456 1.268284722929
O -0.282719799979 -0.968071483919 1.218985745408
H -1.462181879249 2.037574087936 0.413938092372
H -2.089401829129 0.400244944865 -0.213737689812
H -0.004528107436 0.873535072912 2.039546844131
H -0.204997321766 -1.218940513140 0.220256917291
C 0.006214554739 0.388289216687 -1.261214293360
O 0.031387324783 -0.854281954696 -1.181725666911
H 0.876485106222 1.010587986248 -0.955452515649
H -0.680100860291 0.903099900829 -1.965647977365
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005636035079 -0.001755085612 -0.007590644876
C 0.000947362061 -0.001742723936 -0.000448151974
O 0.000042422231 0.000679207593 -0.005373998906
H 0.000474669591 -0.000142382969 -0.000586824066
H -0.000155490577 -0.000142267854 0.000198952601
H -0.000045831275 -0.000127453181 0.000066854981
H 0.000696470936 0.000591272311 -0.000793000272
C -0.006207624949 0.002700437214 0.007679217893
O -0.001042142600 -0.000171665231 0.006904538789
H -0.000302671997 0.000234579302 0.000040450894
H -0.000022157878 -0.000154005045 -0.000104511247
*** Time spent in gradient calculation: 1.68 sec ***
* Info * Energy : -268.1037422258 a.u.
* Info * Gradient : 5.062076e-03 a.u. (RMS)
* Info * 1.023706e-02 a.u. (Max)
* Info * Time : 3.42 sec
Optimization Step 82
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.331480473262 0.955809197833 0.339575670288
C -0.552688801704 0.308787079421 1.268300477923
O -0.282720902373 -0.968089412099 1.219124947769
H -1.462377111766 2.037631961862 0.414181026804
H -2.089336949831 0.400302579097 -0.213820096977
H -0.004508159964 0.873586009042 2.039518942744
H -0.205284165860 -1.219183174838 0.220593822303
C 0.006430097114 0.388195256622 -1.261480756215
O 0.031414375449 -0.854277678331 -1.181905557762
H 0.876611909898 1.010490039273 -0.955469747638
H -0.680090449513 0.903163022727 -1.965606117234
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005649114850 -0.001753272006 -0.007605931608
C 0.000951584863 -0.001754829725 -0.000456970971
O 0.000041828083 0.000686486534 -0.005339076524
H 0.000474374753 -0.000142534313 -0.000590445819
H -0.000155082576 -0.000142802939 0.000198573857
H -0.000046385420 -0.000125465233 0.000066971781
H 0.000695603519 0.000582678780 -0.000826849939
C -0.006216008384 0.002712022938 0.007691921770
O -0.001043702661 -0.000170934518 0.006913202496
H -0.000305929541 0.000234871694 0.000041776433
H -0.000024343202 -0.000156245185 -0.000100536096
*** Time spent in gradient calculation: 1.65 sec ***
* Info * Energy : -268.1037611061 a.u.
* Info * Gradient : 5.067476e-03 a.u. (RMS)
* Info * 1.025473e-02 a.u. (Max)
* Info * Time : 3.46 sec
Optimization Step 83
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.331756977945 0.955895004189 0.339948362302
C -0.552735269073 0.308873192856 1.268323218606
O -0.282722434258 -0.968114981816 1.219319992783
H -1.462652381499 2.037713733092 0.414526090785
H -2.089245607204 0.400384232563 -0.213936120808
H -0.004479581711 0.873656524446 2.039479674766
H -0.205688271262 -1.219520803488 0.221089421521
C 0.006734719543 0.388061967141 -1.261857407539
O 0.031452548073 -0.854271676758 -1.182159735693
H 0.876792807308 1.010351657598 -0.955494974883
H -0.680074190717 0.903253515364 -1.965549565924
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005667517773 -0.001750777557 -0.007628870690
C 0.000958572337 -0.001773010259 -0.000468575613
O 0.000040942009 0.000694514488 -0.005300445891
H 0.000474023227 -0.000142644606 -0.000595431233
H -0.000154657510 -0.000143617345 0.000198112156
H -0.000047229067 -0.000122872007 0.000066735827
H 0.000694515366 0.000574436575 -0.000865041726
C -0.006228856541 0.002729371226 0.007711337545
O -0.001046227822 -0.000171877324 0.006926517417
H -0.000309898768 0.000235602464 0.000043634956
H -0.000027627208 -0.000159067208 -0.000095716773
*** Time spent in gradient calculation: 1.84 sec ***
* Info * Energy : -268.1037878738 a.u.
* Info * Gradient : 5.076814e-03 a.u. (RMS)
* Info * 1.028168e-02 a.u. (Max)
* Info * Time : 3.53 sec
Optimization Step 84
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.332148137900 0.956015521857 0.340475298070
C -0.552801447715 0.308996043525 1.268356088279
O -0.282724506575 -0.968151429684 1.219592655555
H -1.463039651969 2.037828845429 0.415017377707
H -2.089117462496 0.400500244368 -0.214099296115
H -0.004438551789 0.873753504806 2.039424531105
H -0.206256504974 -1.219989842367 0.221821688185
C 0.007164817550 0.387872810842 -1.262389787158
O 0.031506686868 -0.854262935003 -1.182518651382
H 0.877051352939 1.010155509143 -0.955532374520
H -0.680047986839 0.903383676932 -1.965474372780
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005693358537 -0.001747702445 -0.007663357111
C 0.000969820587 -0.001799759746 -0.000483643041
O 0.000039630406 0.000701893501 -0.005263858019
H 0.000473716374 -0.000142686949 -0.000602170685
H -0.000154232017 -0.000144792604 0.000197617010
H -0.000048464479 -0.000119618156 0.000065764035
H 0.000693274590 0.000569110634 -0.000902891884
C -0.006248920841 0.002755037055 0.007740996703
O -0.001050208986 -0.000175840527 0.006947129605
H -0.000314451362 0.000237024843 0.000046213719
H -0.000032420409 -0.000162503750 -0.000090127511
*** Time spent in gradient calculation: 1.87 sec ***
* Info * Energy : -268.1038258614 a.u.
* Info * Gradient : 5.092733e-03 a.u. (RMS)
* Info * 1.032290e-02 a.u. (Max)
* Info * Time : 3.72 sec
Optimization Step 85
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.332701369272 0.956184778493 0.341220705584
C -0.552895872125 0.309171971028 1.268404013438
O -0.282727304553 -0.968203194033 1.219973683454
H -1.463584160865 2.037990830057 0.415717477575
H -2.088937590188 0.400665091015 -0.214328325610
H -0.004379116475 0.873885748214 2.039348622991
H -0.207054481008 -1.220645686635 0.222894801580
C 0.007772312222 0.387603694629 -1.263142283743
O 0.031583158148 -0.854250239260 -1.183025437139
H 0.877420718550 1.009877254111 -0.955588483185
H -0.680004427653 0.903571066270 -1.965375528359
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005729826499 -0.001744324971 -0.007713949860
C 0.000986741804 -0.001836985467 -0.000503238702
O 0.000037772984 0.000706894446 -0.005235378298
H 0.000473623395 -0.000142665456 -0.000611199940
H -0.000153931560 -0.000146584301 0.000196964285
H -0.000050165118 -0.000115753662 0.000063626901
H 0.000692055568 0.000568840597 -0.000935883299
C -0.006279467228 0.002790808097 0.007785756530
O -0.001056373385 -0.000183330518 0.006978343650
H -0.000319542247 0.000239726802 0.000049695405
H -0.000039395625 -0.000166336665 -0.000083947154
*** Time spent in gradient calculation: 1.85 sec ***
* Info * Energy : -268.1038798660 a.u.
* Info * Gradient : 5.118366e-03 a.u. (RMS)
* Info * 1.038452e-02 a.u. (Max)
* Info * Time : 4.37 sec
Optimization Step 86
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.333484018685 0.956421761833 0.342275398398
C -0.553031292341 0.309424770009 1.268474211564
O -0.282730972473 -0.968276155160 1.220506286115
H -1.464348470407 2.038217982599 0.416717352357
H -2.088685482256 0.400900354780 -0.214648915575
H -0.004292706239 0.874064839736 2.039246353115
H -0.208173008584 -1.221570825150 0.224448919690
C 0.008630084795 0.387220319300 -1.264206237420
O 0.031691478684 -0.854231259017 -1.183740753892
H 0.877947946355 1.009480356009 -0.955673730299
H -0.679929990077 0.903840552471 -1.965247327318
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005781065247 -0.001742162429 -0.007786066271
C 0.001009936886 -0.001884140868 -0.000529177840
O 0.000035324458 0.000706433088 -0.005221535697
H 0.000474194534 -0.000142552531 -0.000623042318
H -0.000153641435 -0.000149160156 0.000196000862
H -0.000052140776 -0.000111437514 0.000059984337
H 0.000691380518 0.000574995092 -0.000959840789
C -0.006325004239 0.002836511722 0.007851321937
O -0.001065481971 -0.000191809499 0.007024517861
H -0.000325485571 0.000244105112 0.000054216447
H -0.000048935211 -0.000170341065 -0.000076901328
*** Time spent in gradient calculation: 1.60 sec ***
* Info * Energy : -268.1039567966 a.u.
* Info * Gradient : 5.157308e-03 a.u. (RMS)
* Info * 1.047353e-02 a.u. (Max)
* Info * Time : 3.50 sec
Optimization Step 87
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.334591539565 0.956753404427 0.343768715470
C -0.553225996651 0.309788602739 1.268578112979
O -0.282735679679 -0.968377661822 1.221251572697
H -1.465421154936 2.038535885547 0.418147837773
H -2.088332450526 0.401237385299 -0.215095311744
H -0.004167213018 0.874305810113 2.039113203751
H -0.209739380374 -1.222888362915 0.226664528874
C 0.009841791043 0.386673739980 -1.265711292547
O 0.031844699412 -0.854203420299 -1.184750434932
H 0.878700485699 1.008911282807 -0.955804600114
H -0.679800732196 0.904226476073 -1.965084495398
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005852441895 -0.001743713700 -0.007884740573
C 0.001038501809 -0.001936797731 -0.000564718259
O 0.000032340301 0.000696738921 -0.005225982743
H 0.000476214275 -0.000142716698 -0.000638359574
H -0.000152921354 -0.000152728201 0.000194336711
H -0.000053915883 -0.000107067205 0.000054638526
H 0.000692074145 0.000586172844 -0.000973907000
C -0.006390532471 0.002888584384 0.007944183016
O -0.001078348802 -0.000194638463 0.007090527362
H -0.000333005686 0.000250755115 0.000059919967
H -0.000061522231 -0.000173971184 -0.000068325599
*** Time spent in gradient calculation: 1.58 sec ***
* Info * Energy : -268.1040666841 a.u.
* Info * Gradient : 5.212917e-03 a.u. (RMS)
* Info * 1.059683e-02 a.u. (Max)
* Info * Time : 3.88 sec
Optimization Step 88
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.336159967923 0.957217481006 0.345884157995
C -0.553506189829 0.310310910678 1.268733977411
O -0.282741522696 -0.968516153099 1.222295346752
H -1.466928190312 2.038980581885 0.420199246366
H -2.087841130885 0.401722802900 -0.215711582721
H -0.003987493832 0.874629373377 2.038946926841
H -0.211932197807 -1.224770948635 0.229779134393
C 0.011554203304 0.385895944190 -1.267841891565
O 0.032061567658 -0.854164650452 -1.186175703117
H 0.879776975165 1.008089517422 -0.956007350835
H -0.679574942396 0.904774779965 -1.964886425457
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005949852553 -0.001752759705 -0.008013917794
C 0.001070281316 -0.001986478229 -0.000614904447
O 0.000028840868 0.000672243901 -0.005251419406
H 0.000480897152 -0.000143407375 -0.000657859294
H -0.000150997157 -0.000157689849 0.000191040457
H -0.000054619614 -0.000103158131 0.000047462102
H 0.000695237913 0.000598981923 -0.000978033497
C -0.006481332747 0.002939961739 0.008070329841
O -0.001095646006 -0.000180234788 0.007181713687
H -0.000343374612 0.000259974612 0.000067151353
H -0.000077607178 -0.000176618637 -0.000056710995
*** Time spent in gradient calculation: 1.73 sec ***
* Info * Energy : -268.1042241435 a.u.
* Info * Gradient : 5.288071e-03 a.u. (RMS)
* Info * 1.076017e-02 a.u. (Max)
* Info * Time : 3.96 sec
Optimization Step 89
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.338383749634 0.957868030133 0.348882163882
C -0.553908715731 0.311055007346 1.268972178415
O -0.282748428253 -0.968699491740 1.223758348232
H -1.469052934982 2.039603026964 0.423149727225
H -2.087166042542 0.402427563704 -0.216549005986
H -0.003737902016 0.875065421794 2.038751449459
H -0.215004426010 -1.227449495749 0.234097683270
C 0.013975987128 0.384794602072 -1.270860870587
O 0.032368745706 -0.854116970907 -1.188188399317
H 0.881325085317 1.006894189627 -0.956325503608
H -0.679179871447 0.905544430966 -1.964667552130
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.006077264559 -0.001773070960 -0.008174121771
C 0.001102878934 -0.002022016850 -0.000687193148
O 0.000024461376 0.000625660976 -0.005297839479
H 0.000489777530 -0.000144798767 -0.000682433268
H -0.000146322410 -0.000164611996 0.000184422971
H -0.000053126604 -0.000100149481 0.000038092988
H 0.000701826422 0.000608246749 -0.000972278819
C -0.006600735640 0.002980663104 0.008232565457
O -0.001117624665 -0.000133003863 0.007302437035
H -0.000358210058 0.000271724867 0.000076927394
H -0.000098258428 -0.000177614165 -0.000039560949
*** Time spent in gradient calculation: 2.13 sec ***
* Info * Energy : -268.1044505458 a.u.
* Info * Gradient : 5.383756e-03 a.u. (RMS)
* Info * 1.096491e-02 a.u. (Max)
* Info * Time : 5.78 sec
Optimization Step 90
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.341541210062 0.958783110218 0.353131829482
C -0.554485393060 0.312102151793 1.269345232573
O -0.282755735715 -0.968931123046 1.225809959157
H -1.472068264981 2.040475151230 0.427411327372
H -2.086261526158 0.403462458105 -0.217654822397
H -0.003408655240 0.875657806548 2.038545914776
H -0.219315134351 -1.231213226536 0.240004493538
C 0.017404245778 0.383247334635 -1.275143004969
O 0.032804181807 -0.854073794297 -1.191033173953
H 0.883571060335 1.005143760501 -0.956836801579
H -0.678484367379 0.906607693408 -1.964481727868
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.006231876855 -0.001805727834 -0.008358089599
C 0.001135453764 -0.002032090612 -0.000792433515
O 0.000017855960 0.000546616722 -0.005361659537
H 0.000504343445 -0.000147119917 -0.000713233873
H -0.000136173382 -0.000174165883 0.000171709057
H -0.000048159141 -0.000098017412 0.000025379278
H 0.000712030339 0.000609239746 -0.000952926173
C -0.006746271619 0.003000446936 0.008425567486
O -0.001143526554 -0.000037157774 0.007453165131
H -0.000378934079 0.000285584643 0.000091463546
H -0.000125783985 -0.000176315430 -0.000013272970
*** Time spent in gradient calculation: 1.87 sec ***
* Info * Energy : -268.1047770907 a.u.
* Info * Gradient : 5.496726e-03 a.u. (RMS)
* Info * 1.120290e-02 a.u. (Max)
* Info * Time : 4.89 sec
Optimization Step 91
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.346029762211 0.960075160431 0.359154758238
C -0.555311603766 0.313553118570 1.269947480744
O -0.282760855495 -0.969200876210 1.228688590992
H -1.476387849034 2.041697792098 0.433606670804
H -2.085105124222 0.405004922919 -0.219040529104
H -0.003010567966 0.876469621454 2.038388990163
H -0.225373392360 -1.236414064918 0.247926969873
C 0.022261903162 0.381094204206 -1.281222620568
O 0.033422199552 -0.854072518251 -1.195060485767
H 0.886865243884 1.002563583372 -0.957690719188
H -0.677242362679 0.908047404234 -1.964472134809
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.006395410428 -0.001846856237 -0.008541007881
C 0.001169140781 -0.002005149942 -0.000946427496
O 0.000005618602 0.000426267503 -0.005407839849
H 0.000525132182 -0.000149268969 -0.000751189487
H -0.000116074334 -0.000186793916 0.000148682903
H -0.000038030968 -0.000095515208 0.000007627035
H 0.000724554773 0.000590497354 -0.000931756288
C -0.006900769561 0.002985882913 0.008624992729
O -0.001170045403 0.000124426182 0.007623321495
H -0.000407309776 0.000300365144 0.000114104659
H -0.000163501219 -0.000172055277 0.000027930440
*** Time spent in gradient calculation: 1.79 sec ***
* Info * Energy : -268.1052488032 a.u.
* Info * Gradient : 5.611744e-03 a.u. (RMS)
* Info * 1.144232e-02 a.u. (Max)
* Info * Time : 4.59 sec
Optimization Step 92
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.352408133937 0.961903290772 0.367681746728
C -0.556504307024 0.315533464004 1.270957708406
O -0.282754776309 -0.969471979044 1.232698859156
H -1.482638753716 2.043404785045 0.442705723322
H -2.083764592273 0.407331650360 -0.220611638098
H -0.002605984342 0.877566993172 2.038427630461
H -0.233881300084 -1.243343630464 0.258624544036
C 0.029145195824 0.378125821206 -1.289853794512
O 0.034299551088 -0.854198191062 -1.200774433502
H 0.891780139066 0.998744154451 -0.959182347198
H -0.674983416646 0.909961839563 -1.965005254253
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.006526739435 -0.001873228363 -0.008685021565
C 0.001226385935 -0.001954786421 -0.001157765077
O -0.000021230228 0.000211535271 -0.005548178746
H 0.000550669400 -0.000149014565 -0.000794730728
H -0.000088547740 -0.000207742710 0.000106527083
H -0.000022267066 -0.000092267423 -0.000024287504
H 0.000738074879 0.000606272832 -0.000758105339
C -0.007036201914 0.002946753315 0.008794639474
O -0.001194249975 0.000326707933 0.007795473590
H -0.000434094211 0.000319083089 0.000150083444
H -0.000219234435 -0.000160334866 0.000080563146
*** Time spent in gradient calculation: 1.81 sec ***
* Info * Energy : -268.1059286585 a.u.
* Info * Gradient : 5.719017e-03 a.u. (RMS)
* Info * 1.164204e-02 a.u. (Max)
* Info * Time : 4.54 sec
Optimization Step 93
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.361454527400 0.964515547320 0.379738980618
C -0.558272960061 0.318126240227 1.272733773017
O -0.282704341450 -0.969529580076 1.238527498503
H -1.491952593685 2.045752887958 0.456139017120
H -2.082399499551 0.410982175646 -0.221849904009
H -0.002466586493 0.879180149885 2.039226787528
H -0.246019636031 -1.253534199513 0.268420104217
C 0.038929761612 0.374117007119 -1.302130294340
O 0.035546386384 -0.854652042023 -1.208929775648
H 0.898980322163 0.992924362716 -0.961920691964
H -0.670685624996 0.912174113512 -1.966668329003
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.006453719936 -0.001938986873 -0.008517554342
C 0.001120025514 -0.001621645559 -0.001564467874
O -0.000036111853 0.000644099834 -0.002935666384
H 0.000570045890 -0.000158440249 -0.000850898484
H 0.000051044810 -0.000165537139 0.000095810858
H 0.000028721101 -0.000052427825 0.000009469680
H 0.000700449690 -0.000402176676 -0.003083473993
C -0.006912496104 0.002664617131 0.008596417125
O -0.001145652386 0.000930261244 0.007750909391
H -0.000584114390 0.000276420034 0.000174077763
H -0.000218818582 -0.000199794943 0.000280753607
*** Time spent in gradient calculation: 1.87 sec ***
* Info * Energy : -268.1068883097 a.u.
* Info * Gradient : 5.528869e-03 a.u. (RMS)
* Info * 1.134818e-02 a.u. (Max)
* Info * Time : 4.51 sec
Optimization Step 94
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.373300918913 0.967627310647 0.395618954031
C -0.560558437439 0.321830075893 1.274849378933
O -0.282762631118 -0.969983656920 1.245492350508
H -1.503101731382 2.048715591348 0.473972998681
H -2.080758754309 0.416628153653 -0.224240418095
H -0.002188899903 0.880790304516 2.039700432220
H -0.261913083787 -1.264099943148 0.294300627435
C 0.051686355646 0.368544027665 -1.318177764449
O 0.037134989083 -0.854989403259 -1.219561626087
H 0.908815944198 0.985432143384 -0.965382399118
H -0.665624921906 0.915877642701 -1.968576471684
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.006561412637 -0.002052351787 -0.008387536812
C 0.001380446382 -0.001305604574 -0.002004795799
O -0.000214179202 -0.001640916265 -0.008581637259
H 0.000615400001 -0.000133775025 -0.000900746709
H 0.000051080786 -0.000196607019 -0.000118284015
H 0.000003853298 -0.000117779468 -0.000213677323
H 0.000884288663 0.001589583069 0.002669459839
C -0.006907134758 0.002042349632 0.008747700570
O -0.001307326250 0.001467646739 0.008113983184
H -0.000542290048 0.000374424981 0.000357807243
H -0.000508118141 -0.000043218645 0.000275138216
*** Time spent in gradient calculation: 1.93 sec ***
* Info * Energy : -268.1081387481 a.u.
* Info * Gradient : 6.122869e-03 a.u. (RMS)
* Info * 1.133146e-02 a.u. (Max)
* Info * Time : 4.53 sec
Optimization Step 95
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.385671304658 0.971150432176 0.412084310504
C -0.563234741143 0.325103717481 1.278042041070
O -0.282393682809 -0.969247886249 1.253802336958
H -1.515700740671 2.051649163102 0.494986710478
H -2.080225240906 0.422313696603 -0.223569322195
H -0.002438247294 0.883031832361 2.042226636636
H -0.278784386287 -1.279433592696 0.296740154949
C 0.064933787657 0.363447608561 -1.335195965566
O 0.038921528525 -0.856103047276 -1.230984633587
H 0.920334250949 0.975205681258 -0.970917242088
H -0.656594329305 0.918352038656 -1.973383705820
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005941606258 -0.002164611135 -0.007347683745
C 0.000702245717 -0.000048159178 -0.002861225296
O 0.000014379153 0.001011482212 0.001837773147
H 0.000730075185 -0.000193695074 -0.000836531739
H 0.000397442502 -0.000146747316 -0.000128933320
H 0.000194615138 -0.000005124068 0.000004814862
H 0.000572289411 -0.002510171560 -0.007005862501
C -0.006522083056 0.001060332044 0.007427096555
O -0.000919011718 0.003115925216 0.007649684315
H -0.000811700045 0.000080646636 0.000425207783
H -0.000289937854 -0.000211696792 0.000807379866
*** Time spent in gradient calculation: 1.85 sec ***
* Info * Energy : -268.1093538901 a.u.
* Info * Gradient : 5.508639e-03 a.u. (RMS)
* Info * 9.941008e-03 a.u. (Max)
* Info * Time : 4.25 sec
Optimization Step 96
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.395312612313 0.973960492147 0.425132116425
C -0.565572158879 0.327493284680 1.281403155633
O -0.282346451349 -0.969056131416 1.258478340804
H -1.527020085110 2.054090471190 0.511628435008
H -2.081830413597 0.427345872756 -0.222035086022
H -0.004202917814 0.883736099057 2.044847843315
H -0.290988763354 -1.280407727444 0.326802579276
C 0.075703292268 0.359322105554 -1.348611165814
O 0.040162323795 -0.857465936805 -1.240288104765
H 0.930163968656 0.969265020888 -0.976030216928
H -0.649389324521 0.920845250002 -1.979898503099
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005616355221 -0.001570799589 -0.007496828496
C 0.001755164008 -0.001212469349 -0.002643444285
O -0.000685486115 -0.003795457056 -0.011905490141
H 0.000614782702 -0.000064516675 -0.000871057438
H 0.000141491443 -0.000278070603 -0.000315388612
H 0.000055899999 -0.000071341159 -0.000402852046
H 0.001179476981 0.003084184153 0.007094102928
C -0.006163758445 0.001988676239 0.007698217942
O -0.001250311966 0.001476557769 0.007875213311
H -0.000525215532 0.000366164915 0.000565418531
H -0.000743266780 0.000077861953 0.000402610938
*** Time spent in gradient calculation: 1.86 sec ***
* Info * Energy : -268.1103179582 a.u.
* Info * Gradient : 6.671021e-03 a.u. (RMS)
* Info * 1.251463e-02 a.u. (Max)
* Info * Time : 4.44 sec
Optimization Step 97
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.403898247547 0.976576287756 0.436947333412
C -0.567983434459 0.329578323004 1.285171511524
O -0.281731317636 -0.967667414157 1.264306483739
H -1.537968639567 2.055936673383 0.527808279110
H -2.083226587881 0.431841091507 -0.219068207050
H -0.006445863771 0.884656821342 2.049906806190
H -0.303350955605 -1.291133393988 0.320694719815
C 0.085409364431 0.355785736863 -1.360830450616
O 0.041371004027 -0.858990364720 -1.249069863751
H 0.939263348916 0.962393488531 -0.982377904660
H -0.640038377898 0.921500748373 -1.986867006974
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.005005306166 -0.001599857525 -0.006580486358
C 0.001310699510 -0.000329417303 -0.003042229743
O -0.000155029182 0.000383290599 0.001220111802
H 0.000601735822 -0.000106723827 -0.000855960375
H 0.000312647668 -0.000168008951 -0.000246657241
H 0.000180273917 -0.000007212361 -0.000136547533
H 0.000521208172 -0.002106043747 -0.005488568106
C -0.005609074182 0.001319418171 0.006712166892
O -0.000988006370 0.002366136250 0.007199516624
H -0.000721957004 0.000331718668 0.000474613734
H -0.000463552396 -0.000086620238 0.000742894349
*** Time spent in gradient calculation: 1.91 sec ***
* Info * Energy : -268.1112039164 a.u.
* Info * Gradient : 4.844178e-03 a.u. (RMS)
* Info * 8.846229e-03 a.u. (Max)
* Info * Time : 4.21 sec
Optimization Step 98
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.413116709086 0.979329327395 0.449871283361
C -0.571031551147 0.331751654025 1.289949402385
O -0.281422589715 -0.966614719958 1.268891999455
H -1.550132733447 2.057956762380 0.545647046268
H -2.085749390560 0.436690882508 -0.215395275669
H -0.010176808744 0.884878736394 2.055713591227
H -0.316110911982 -1.291893291812 0.342507710092
C 0.096147700319 0.351712837614 -1.374227921813
O 0.042600651690 -0.860845646304 -1.258883087931
H 0.949930802986 0.955389840533 -0.989250747252
H -0.630240385515 0.922931229853 -1.996062046958
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.004507731138 -0.001085704641 -0.006459970060
C 0.001999366781 -0.001156164678 -0.002913191975
O -0.000837124056 -0.002882744706 -0.007652591760
H 0.000498393685 -0.000023366617 -0.000855562738
H 0.000134679401 -0.000244241331 -0.000306475188
H 0.000113091371 -0.000027277576 -0.000325154409
H 0.001086374381 0.001679665015 0.003850585182
C -0.005083180234 0.002025805794 0.006662584466
O -0.001161864366 0.001177350641 0.007036040836
H -0.000574436586 0.000485375919 0.000488015028
H -0.000689571706 0.000051783631 0.000486540528
*** Time spent in gradient calculation: 1.84 sec ***
* Info * Energy : -268.1121121574 a.u.
* Info * Gradient : 5.159433e-03 a.u. (RMS)
* Info * 8.621638e-03 a.u. (Max)
* Info * Time : 4.34 sec
Optimization Step 99
======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.422335743895 0.982016926491 0.463294555398
C -0.574551454365 0.334262494652 1.295318218570
O -0.280457531944 -0.964719556570 1.274514887401
H -1.562486915287 2.059615112673 0.564840340602
H -2.087188378259 0.441908038232 -0.211719845662
H -0.014743411742 0.885186623260 2.063510700321
H -0.329910148574 -1.300214685230 0.343565098917
C 0.107156170509 0.347240064847 -1.388286873428
O 0.043876999039 -0.862606815343 -1.269167353254
H 0.960815877751 0.946524061534 -0.996337184354
H -0.618849296247 0.923905959992 -2.005278350026
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.004104751923 -0.001227780200 -0.005558933176
C 0.001603244812 -0.000267266949 -0.003228770318
O -0.000348043715 -0.000216073123 0.000429756359
H 0.000504718604 -0.000056900419 -0.000810312499
H 0.000231906065 -0.000166884721 -0.000258479831
H 0.000170817376 -0.000031133441 -0.000187987221
H 0.000495277976 -0.001673494574 -0.003874516543
C -0.004621850147 0.001009499339 0.005774846695
O -0.000904367356 0.002233446993 0.006504986437
H -0.000686929815 0.000415062155 0.000494476179
H -0.000550355053 -0.000023783091 0.000716079181
*** Time spent in gradient calculation: 1.70 sec ***
* Info * Energy : -268.1129799863 a.u.
* Info * Gradient : 4.127723e-03 a.u. (RMS)
* Info * 7.465215e-03 a.u. (Max)
* Info * Time : 3.93 sec
Optimization Step 100
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.431439122651 0.984808071928 0.476405838103
C -0.578444713532 0.336098173629 1.301912366666
O -0.279767400037 -0.962750466757 1.279114791265
H -1.576126698396 2.061245080440 0.584407380943
H -2.090728559754 0.446882619212 -0.206387763539
H -0.020428130861 0.885158194754 2.072146581300
H -0.344358508242 -1.299284447669 0.360425162977
C 0.118333024347 0.343332224337 -1.402062794312
O 0.045008583146 -0.865235178261 -1.279937647705
H 0.973282943560 0.938872297962 -1.005783552724
H -0.606054968932 0.924763541782 -2.017979715584
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003401451047 -0.000603455980 -0.005499728508
C 0.002317168064 -0.001390580288 -0.002869495585
O -0.000985830535 -0.002317421091 -0.005437912465
H 0.000375591241 0.000021334984 -0.000786969665
H 0.000040935961 -0.000215036935 -0.000255105542
H 0.000122122980 -0.000020615213 -0.000308218303
H 0.000955593437 0.001135270045 0.002570573199
C -0.004004116921 0.002417094611 0.005698641335
O -0.001052539042 0.000279261121 0.006093587882
H -0.000494056531 0.000615571407 0.000377439621
H -0.000667083604 0.000070950898 0.000412082475
*** Time spent in gradient calculation: 1.86 sec ***
* Info * Energy : -268.1138126644 a.u.
* Info * Gradient : 4.237045e-03 a.u. (RMS)
* Info * 7.372232e-03 a.u. (Max)
* Info * Time : 4.12 sec
Optimization Step 101
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.440310582344 0.987266790555 0.490057189948
C -0.583050700266 0.338491519020 1.309255295573
O -0.278378654317 -0.960072031607 1.284174788388
H -1.589628427585 2.062234051657 0.605356872349
H -2.092995639132 0.451998215116 -0.201153893265
H -0.026963836961 0.885073211097 2.082546621402
H -0.359920486577 -1.303228911350 0.364476191652
C 0.129742518107 0.338522143955 -1.416524740129
O 0.046187733282 -0.867384325355 -1.291214447290
H 0.986105188950 0.928538095294 -1.014939963336
H -0.591779293791 0.925423045008 -2.030667772378
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003130663808 -0.000762070868 -0.004852815143
C 0.001955946746 -0.000935693958 -0.002925139128
O -0.000519387169 -0.000461949036 -0.000399840627
H 0.000368442617 -0.000012931248 -0.000701806758
H 0.000041652566 -0.000145292300 -0.000151492483
H 0.000169221297 -0.000033195388 -0.000250557703
H 0.000368030357 -0.000829262599 -0.002170726148
C -0.003751536637 0.001542820406 0.005209076387
O -0.000740626076 0.001124752069 0.005476124879
H -0.000508402334 0.000504132955 0.000279471975
H -0.000497007909 -0.000004667849 0.000466975318
*** Time spent in gradient calculation: 1.86 sec ***
* Info * Energy : -268.1146084153 a.u.
* Info * Gradient : 3.445425e-03 a.u. (RMS)
* Info * 6.602181e-03 a.u. (Max)
* Info * Time : 4.38 sec
Optimization Step 102
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.449903048012 0.989660061810 0.504992042471
C -0.588662132529 0.341419418349 1.317375964337
O -0.277106733945 -0.957557708451 1.289185739298
H -1.602811924302 2.063127724088 0.628330554656
H -2.093690026959 0.457843732243 -0.197225258135
H -0.034651940777 0.884884403967 2.093844825924
H -0.376487238595 -1.305362487398 0.380270334579
C 0.142089418718 0.332933721372 -1.432695287357
O 0.047377614894 -0.869349852977 -1.303403498757
H 1.000147309011 0.915246046959 -1.023083717889
H -0.577013055501 0.927216048907 -2.043252385064
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.003056631298 -0.001106016360 -0.004081623615
C 0.001347469404 0.000034718392 -0.003163371802
O -0.000949243258 -0.002285680604 -0.004118043198
H 0.000470618605 -0.000056161034 -0.000549523982
H 0.000102732867 -0.000134525090 -0.000147031453
H 0.000216808275 -0.000047117868 -0.000315747749
H 0.000819746094 0.000645830224 0.001756434235
C -0.003738112590 0.000320432365 0.004410052594
O -0.000304327479 0.002466504734 0.005192313284
H -0.000490733944 0.000141027302 0.000436223398
H -0.000506562242 0.000008342264 0.000554892944
*** Time spent in gradient calculation: 1.74 sec ***
* Info * Energy : -268.1153931144 a.u.
* Info * Gradient : 3.499481e-03 a.u. (RMS)
* Info * 5.790054e-03 a.u. (Max)
* Info * Time : 4.42 sec
Optimization Step 103
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.458283831677 0.992902604875 0.517368141666
C -0.593349979948 0.341795234274 1.327935925138
O -0.275315510118 -0.953623034011 1.293631351024
H -1.622134247131 2.063979203201 0.649214724721
H -2.099708006060 0.461503109920 -0.188216476953
H -0.043573674369 0.884838317450 2.107691547996
H -0.394242995981 -1.299420150956 0.383172927906
C 0.153844254910 0.330535536555 -1.445778664895
O 0.047925858295 -0.874473793183 -1.315826381478
H 1.016322292267 0.909960901318 -1.039547548796
H -0.557653189975 0.925898116142 -2.064955860624
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.001487978823 0.000625663162 -0.005108669513
C 0.003283788653 -0.003547849391 -0.001420143099
O -0.000566684825 0.000089155632 -0.000383007792
H 0.000051759706 0.000123586028 -0.000601014799
H -0.000298855082 -0.000189126830 -0.000026456362
H 0.000151179282 0.000110922338 -0.000360772368
H -0.000034517808 0.000182919587 -0.001171590214
C -0.002735104381 0.006105785382 0.005180432867
O -0.000998192393 -0.004453041292 0.004306304465
H 0.000050426382 0.000943239299 -0.000238464528
H -0.000367955485 -0.000006128082 -0.000205195292
*** Time spent in gradient calculation: 1.65 sec ***
* Info * Energy : -268.1160316589 a.u.
* Info * Gradient : 3.916629e-03 a.u. (RMS)
* Info * 8.461578e-03 a.u. (Max)
* Info * Time : 3.70 sec
Optimization Step 104
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.467315522736 0.994026275886 0.532278541059
C -0.599475048841 0.346310748839 1.333586367122
O -0.273691789256 -0.952037891962 1.299306664787
H -1.628294494393 2.064289117366 0.671013757546
H -2.092949346248 0.469479195272 -0.191478382014
H -0.051286726808 0.884547245532 2.117439981544
H -0.407819313268 -1.313171945776 0.393997609562
C 0.165129518577 0.322742267880 -1.462664464776
O 0.049117091525 -0.873151096039 -1.326287591657
H 1.026707076134 0.890832032088 -1.040164431417
H -0.547198763448 0.930156321968 -2.068502712199
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.002688190486 -0.001733183042 -0.002492516189
C 0.000197697457 0.001911297610 -0.003541796249
O 0.000170567303 -0.000191479805 0.002666112870
H 0.000638360914 -0.000107852986 -0.000306562193
H 0.000343233242 -0.000032699154 -0.000185078449
H 0.000305900533 -0.000017385505 -0.000295016610
H -0.000326826786 -0.002392085207 -0.004648364658
C -0.003739504065 -0.002339657993 0.002761457999
O 0.000653524461 0.005303273663 0.004473278671
H -0.000559977567 -0.000381287356 0.000653840540
H -0.000351475449 -0.000028549143 0.000888495191
*** Time spent in gradient calculation: 1.70 sec ***
* Info * Energy : -268.1166118733 a.u.
* Info * Gradient : 3.637962e-03 a.u. (RMS)
* Info * 6.968646e-03 a.u. (Max)
* Info * Time : 3.93 sec
Optimization Step 105
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.471663036536 0.996134766747 0.540309211864
C -0.603558504888 0.347154703431 1.341961689056
O -0.272632658315 -0.949129056375 1.300407814503
H -1.643452807781 2.064261530226 0.683135919847
H -2.096915460815 0.470329234888 -0.183606055946
H -0.060758001676 0.881867040096 2.131578717276
H -0.414669081616 -1.304502239309 0.408939633396
C 0.172940050911 0.319828289742 -1.470772561371
O 0.049080486440 -0.876163262904 -1.335072292241
H 1.036007648564 0.887699058219 -1.049987026546
H -0.533384405792 0.928048523474 -2.084233264119
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.002501030911 -0.000723898599 -0.003087451009
C 0.001203927669 0.000021154354 -0.002559486506
O -0.001694531256 -0.003783682358 -0.007872474572
H 0.000215026344 -0.000041598545 -0.000509230025
H -0.000150027517 -0.000091283319 0.000015594275
H 0.000202032734 -0.000090414794 -0.000252183738
H 0.001387059608 0.002418218748 0.006108078604
C -0.002962803019 -0.000559615785 0.004142639506
O 0.000036874289 0.002670750159 0.003669734390
H -0.000370884238 0.000228465873 0.000063923058
H -0.000347192263 -0.000048843423 0.000274883312
*** Time spent in gradient calculation: 1.93 sec ***
* Info * Energy : -268.1169978765 a.u.
* Info * Gradient : 4.223889e-03 a.u. (RMS)
* Info * 8.897390e-03 a.u. (Max)
* Info * Time : 4.40 sec
Optimization Step 106
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.477549028777 0.997739135656 0.547465924007
C -0.605971919451 0.346968469588 1.346565648680
O -0.271213550388 -0.947267540634 1.304574409422
H -1.650239175937 2.065017470213 0.696387178950
H -2.096530449667 0.474817843264 -0.184104360965
H -0.065441585599 0.883451175123 2.136663722097
H -0.425265771940 -1.306142257038 0.404583416560
C 0.180010168511 0.319498696362 -1.479552763910
O 0.049019279993 -0.878947818843 -1.341397429602
H 1.045953841360 0.882232266912 -1.056702915822
H -0.524206056656 0.930034934692 -2.093063159581
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000881611775 0.000196662707 -0.003496793882
C 0.002417967924 -0.001740671586 -0.001860485576
O -0.000042990410 0.000441275391 0.001943391966
H 0.000294299955 0.000083183051 -0.000357591385
H 0.000002159443 -0.000146116853 -0.000173248700
H 0.000163131750 0.000097465156 -0.000350644662
H -0.000537246817 -0.001156898578 -0.003085948072
C -0.002313806281 0.004725171758 0.002891671215
O -0.000380376018 -0.002984248160 0.004039190189
H 0.000028054278 0.000402463009 0.000256491925
H -0.000497892177 0.000075426285 0.000177140018
*** Time spent in gradient calculation: 1.71 sec ***
* Info * Energy : -268.1173195112 a.u.
* Info * Gradient : 3.057902e-03 a.u. (RMS)
* Info * 6.003558e-03 a.u. (Max)
* Info * Time : 3.92 sec
Optimization Step 107
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.482304916826 0.998828537593 0.557306859749
C -0.611382840541 0.349657710948 1.354061046371
O -0.269859537194 -0.944734852331 1.306741155913
H -1.661697002926 2.064234956178 0.710464459472
H -2.095532806403 0.477589924445 -0.180130504592
H -0.075103224903 0.880874206492 2.150395998436
H -0.430922674117 -1.306321898618 0.415280256098
C 0.188303710044 0.313278278521 -1.489784481395
O 0.049182923526 -0.879033188179 -1.350454153728
H 1.053678391148 0.872282145368 -1.061636790597
H -0.510789057969 0.929460032263 -2.103738799625
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.002176944037 -0.000846778022 -0.002362337954
C 0.000821299771 0.000434015918 -0.002618521419
O -0.000724318415 -0.001605296727 -0.002313173302
H 0.000247535216 -0.000036849482 -0.000408985532
H -0.000050318457 -0.000031622524 0.000009791349
H 0.000224081150 -0.000062607215 -0.000201646073
H 0.000338094577 0.000104967366 0.000844534296
C -0.002792965517 -0.001668071208 0.003419626038
O 0.000442938808 0.003671072519 0.003134612125
H -0.000418918561 0.000117709328 0.000069634767
H -0.000249754272 -0.000076534608 0.000419498716
*** Time spent in gradient calculation: 1.91 sec ***
* Info * Energy : -268.1176983924 a.u.
* Info * Gradient : 2.604013e-03 a.u. (RMS)
* Info * 4.847552e-03 a.u. (Max)
* Info * Time : 4.35 sec
Optimization Step 108
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.491075625036 1.001102053189 0.569860905811
C -0.617088269991 0.350604970788 1.363846806446
O -0.267462264402 -0.941131989296 1.311576370455
H -1.675994876816 2.064244805369 0.732301277489
H -2.095019351845 0.483461967490 -0.178063750377
H -0.086495054474 0.880944035533 2.164349540749
H -0.441764991359 -1.303057302826 0.423181631956
C 0.200758663537 0.310325784222 -1.504614291660
O 0.048612257272 -0.882820342022 -1.362218591467
H 1.069146534669 0.860974444244 -1.070652831133
H -0.492476565111 0.931561594938 -2.120689285848
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000862757256 0.000031594052 -0.002700133241
C 0.001830362817 -0.001056226794 -0.001852989247
O -0.000680931037 -0.001139689181 -0.001648697829
H 0.000274410487 0.000035745163 -0.000272690537
H -0.000022223776 -0.000092923207 -0.000114459008
H 0.000174519035 0.000052925251 -0.000318211473
H 0.000101228044 0.000305826006 0.000751381298
C -0.001925774954 0.003279892514 0.002229499970
O -0.000060438047 -0.001702851888 0.003408041135
H -0.000055039021 0.000224322221 0.000256495694
H -0.000485192663 0.000062273252 0.000254189383
*** Time spent in gradient calculation: 1.76 sec ***
* Info * Energy : -268.1181494873 a.u.
* Info * Gradient : 2.251674e-03 a.u. (RMS)
* Info * 4.408738e-03 a.u. (Max)
* Info * Time : 3.93 sec
Optimization Step 109
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.496798288590 1.002784125643 0.582274456593
C -0.624622964883 0.353482743330 1.376283723848
O -0.264856280524 -0.936221392204 1.313988296782
H -1.693927906154 2.062831987954 0.750104282507
H -2.090920546054 0.485022955865 -0.173382091272
H -0.104170101983 0.876597155016 2.188101816797
H -0.446278136982 -1.301652779384 0.422781920942
C 0.212006333849 0.303240754286 -1.517697191215
O 0.047844578427 -0.884775596638 -1.374878102544
H 1.080229647813 0.848084147285 -1.076352492791
H -0.471398022180 0.930349310242 -2.138916017831
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.001849634459 -0.000740751310 -0.001846911964
C 0.000834767567 0.000570151676 -0.001852333424
O 0.000184850033 0.000895121421 0.002364498712
H 0.000179240266 0.000015202948 -0.000386050007
H -0.000124852770 -0.000103256390 0.000134878367
H 0.000121662169 -0.000102921054 -0.000062549392
H -0.000867817047 -0.002139509684 -0.003882601772
C -0.002336285767 -0.001294167827 0.002729684556
O 0.000698305780 0.002846241651 0.002402615796
H -0.000255008513 0.000144702483 0.000020697852
H -0.000271361916 -0.000090696339 0.000361535145
*** Time spent in gradient calculation: 1.83 sec ***
* Info * Energy : -268.1185380451 a.u.
* Info * Gradient : 2.489343e-03 a.u. (RMS)
* Info * 4.517212e-03 a.u. (Max)
* Info * Time : 4.02 sec
Optimization Step 110
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.501788129961 1.003818891641 0.588969253493
C -0.628109420386 0.353779016027 1.381164685310
O -0.263502437649 -0.934658852081 1.316811226953
H -1.700743640103 2.062472181898 0.763072218306
H -2.090456616513 0.489911041178 -0.173625900727
H -0.109183900810 0.877742247290 2.193316308556
H -0.447508152543 -1.295842094359 0.432897082620
C 0.219095957842 0.301266797650 -1.525674318895
O 0.046969494004 -0.886235820450 -1.381217594796
H 1.088374057670 0.841778788687 -1.081057057705
H -0.459471958611 0.931596377018 -2.149290024092
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.001198932933 -0.000290283678 -0.001986318235
C 0.001017136557 -0.000311852505 -0.001791566917
O -0.001006802017 -0.001737698543 -0.003146046368
H 0.000220169748 -0.000013772913 -0.000242080928
H -0.000029663342 -0.000063539906 0.000010289471
H 0.000236263866 0.000039761422 -0.000241153913
H 0.000384215205 0.000865550626 0.002236681852
C -0.001897573342 0.000463387967 0.002409134591
O 0.000445534186 0.000878648006 0.002448663197
H -0.000230136882 0.000181557607 -0.000010291643
H -0.000320915882 -0.000007668528 0.000302262211
*** Time spent in gradient calculation: 1.83 sec ***
* Info * Energy : -268.1187611183 a.u.
* Info * Gradient : 2.061185e-03 a.u. (RMS)
* Info * 3.732406e-03 a.u. (Max)
* Info * Time : 4.12 sec
Optimization Step 111
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.506915450691 1.004974674204 0.597187573736
C -0.632891704512 0.354806175024 1.388527572078
O -0.261348447078 -0.931780851777 1.319785807240
H -1.711456696787 2.061685936010 0.776771728218
H -2.088381314024 0.493651992392 -0.172788930312
H -0.119589265039 0.876676988272 2.205507722885
H -0.448786966238 -1.293962401724 0.432216252730
C 0.227632843330 0.297811514645 -1.535146003468
O 0.045635683480 -0.887866162046 -1.389400094153
H 1.097349952227 0.832654838060 -1.084134657245
H -0.443629463006 0.932051612225 -2.162817246455
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.001078089884 -0.000296548834 -0.001743662716
C 0.000959672324 -0.000200656939 -0.001617238442
O -0.000175994795 -0.000141066493 0.000711511728
H 0.000208604435 0.000018113955 -0.000218917128
H -0.000041488329 -0.000045783639 -0.000027990586
H 0.000222188744 0.000033024652 -0.000266076626
H -0.000498188196 -0.000765351078 -0.001536224378
C -0.001812535700 0.000484541394 0.002044711857
O 0.000562544121 0.000748191792 0.002262181667
H -0.000133090583 0.000178165151 0.000043489260
H -0.000352837879 -0.000011939160 0.000326939829
*** Time spent in gradient calculation: 1.81 sec ***
* Info * Energy : -268.1190065538 a.u.
* Info * Gradient : 1.531887e-03 a.u. (RMS)
* Info * 2.775052e-03 a.u. (Max)
* Info * Time : 4.05 sec
Optimization Step 112
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.512358082550 1.006212741927 0.606242888546
C -0.638612843195 0.356036773953 1.397707766413
O -0.258822434341 -0.928193698380 1.322325022310
H -1.724488898387 2.060362095009 0.791853137592
H -2.085247891304 0.496881866639 -0.171537168901
H -0.133928605507 0.874277616396 2.222247462036
H -0.446989269640 -1.287878476449 0.437369484454
C 0.237545434885 0.293781839560 -1.545818860882
O 0.043567276834 -0.889962594900 -1.398829982646
H 1.107100641367 0.821576343438 -1.086296446623
H -0.423897848181 0.932727676942 -2.179382529636
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000862685881 -0.000158435562 -0.001724067019
C 0.001130080201 -0.000265772479 -0.001373976300
O -0.000762749831 -0.001059676054 -0.001653730420
H 0.000199724114 0.000021109044 -0.000222179530
H -0.000091245203 -0.000064938461 0.000021646210
H 0.000177466428 0.000012049964 -0.000198632605
H 0.000016046670 0.000260179579 0.000901352185
C -0.001577779332 0.001178845359 0.001666865039
O 0.000572602817 -0.000056002944 0.002175913393
H -0.000102677655 0.000104870849 0.000061343588
H -0.000404458000 0.000029638989 0.000323641772
*** Time spent in gradient calculation: 1.89 sec ***
* Info * Energy : -268.1192726122 a.u.
* Info * Gradient : 1.492058e-03 a.u. (RMS)
* Info * 2.580214e-03 a.u. (Max)
* Info * Time : 4.16 sec
Optimization Step 113
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.519196314533 1.007421754819 0.617122460776
C -0.645828604855 0.357223136040 1.407940647383
O -0.255331807380 -0.924057735470 1.326200356985
H -1.739890282863 2.058306417715 0.810973914767
H -2.080641984681 0.502179611418 -0.171640599503
H -0.149727684604 0.872655541928 2.239437120181
H -0.441975799103 -1.281823059354 0.438598688758
C 0.249840402245 0.288351077115 -1.558038299637
O 0.040299853434 -0.891567555176 -1.410160609873
H 1.118366510284 0.809463317964 -1.088939314034
H -0.396783395966 0.932269764612 -2.202103110286
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000879741416 -0.000226697794 -0.001292259259
C 0.000723415143 -0.000077187525 -0.001487308577
O -0.000484933790 -0.000592403876 -0.000120857677
H 0.000191901877 0.000011584572 -0.000162151845
H -0.000031546976 -0.000033418788 -0.000002348187
H 0.000248308148 0.000058225921 -0.000243273700
H -0.000272933000 -0.000247971217 -0.000418002084
C -0.001576174407 -0.000083379004 0.002006048627
O 0.000787719270 0.000948878441 0.001566354482
H -0.000109283517 0.000240740148 -0.000168656370
H -0.000336745335 0.000001548411 0.000294062375
*** Time spent in gradient calculation: 1.95 sec ***
* Info * Energy : -268.1195687273 a.u.
* Info * Gradient : 1.247752e-03 a.u. (RMS)
* Info * 2.552549e-03 a.u. (Max)
* Info * Time : 4.92 sec
Optimization Step 114
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.525208943384 1.008824547855 0.627590555187
C -0.653995725500 0.358274453247 1.421139221447
O -0.250741418122 -0.918331409269 1.328481864690
H -1.759201026793 2.055436526412 0.828982981736
H -2.073104386811 0.505404532407 -0.171975609001
H -0.174282573688 0.867021743412 2.266138586657
H -0.431420090083 -1.271321430669 0.437822678969
C 0.263168407532 0.283200636909 -1.571447110098
O 0.035465832198 -0.894502570120 -1.421693526119
H 1.128264707532 0.792143845680 -1.083199832115
H -0.365222605363 0.932861698113 -2.227557501259
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000537116322 0.000092218962 -0.001535593209
C 0.001429321322 -0.000415880914 -0.000760245536
O -0.000762400851 -0.000209708812 -0.000315611827
H 0.000141620096 0.000065316360 -0.000246901407
H -0.000174601753 -0.000125486295 0.000094454708
H 0.000089638503 -0.000030616371 -0.000118648891
H -0.000259158319 -0.000326509100 -0.000351512578
C -0.001229319023 0.002511711926 0.000668784844
O 0.000722461214 -0.001529352388 0.001973912126
H 0.000049566333 -0.000115372230 0.000166592125
H -0.000527986914 0.000082314232 0.000394331280
*** Time spent in gradient calculation: 2.06 sec ***
* Info * Energy : -268.1198737978 a.u.
* Info * Gradient : 1.418820e-03 a.u. (RMS)
* Info * 2.875273e-03 a.u. (Max)
* Info * Time : 4.89 sec
Optimization Step 115
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.532491850589 1.009411625797 0.637764843585
C -0.661118490066 0.359062085661 1.429437138920
O -0.246517165081 -0.914888748102 1.333019152311
H -1.772730310564 2.052635756668 0.848918409685
H -2.068493205326 0.513414702451 -0.174338911579
H -0.187996837590 0.866905235705 2.278750344669
H -0.420815209905 -1.263959104898 0.441843405177
C 0.275871310802 0.277269219398 -1.582710734216
O 0.030614143289 -0.894486250165 -1.432174271101
H 1.136701126024 0.782005529805 -1.082815780013
H -0.331796581986 0.930403814889 -2.255247887536
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000671651820 -0.000188140548 -0.000823083869
C 0.000454910394 0.000134073098 -0.001506249395
O -0.000967024034 -0.001491646559 -0.001444182502
H 0.000193280924 0.000008287412 -0.000085186442
H -0.000004803875 0.000017841255 -0.000039417602
H 0.000307605333 0.000099875810 -0.000281829327
H 0.000164659848 0.000555957977 0.001345737758
C -0.001496283054 -0.000737123807 0.001822733654
O 0.001153924932 0.001360200279 0.000956912987
H -0.000080758279 0.000205390396 -0.000317579340
H -0.000383031792 0.000036309005 0.000344770273
*** Time spent in gradient calculation: 1.94 sec ***
* Info * Energy : -268.1201299219 a.u.
* Info * Gradient : 1.410987e-03 a.u. (RMS)
* Info * 2.470743e-03 a.u. (Max)
* Info * Time : 4.60 sec
Optimization Step 116
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.536195698761 1.009959752128 0.643801105024
C -0.666637953334 0.359305409279 1.437781252215
O -0.242746640368 -0.910854901728 1.334312844783
H -1.785855965546 2.049933737944 0.860101191878
H -2.062763015301 0.515477532281 -0.175469762663
H -0.205600459699 0.862348742750 2.296504836169
H -0.412292865767 -1.257301669205 0.438302381577
C 0.284928343225 0.273792897109 -1.590410230287
O 0.026037187801 -0.895921097579 -1.439168672540
H 1.140998888599 0.771584797229 -1.075553584313
H -0.305983447696 0.929262863482 -2.275441421863
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000463470018 0.000022646046 -0.001038507831
C 0.000974093530 -0.000130050655 -0.001034695250
O -0.000729655894 -0.000292467074 0.000607098189
H 0.000168924955 0.000044967649 -0.000142322586
H -0.000110256640 -0.000052142940 0.000030312551
H 0.000185263377 0.000026481742 -0.000188102084
H -0.000281214704 -0.000417309746 -0.000800041627
C -0.001251300465 0.000869921942 0.001256646553
O 0.001002465043 -0.000264166694 0.001113449181
H 0.000077935257 0.000112422286 -0.000184751014
H -0.000491246338 0.000079969645 0.000350816879
*** Time spent in gradient calculation: 1.88 sec ***
* Info * Energy : -268.1203247694 a.u.
* Info * Gradient : 1.043891e-03 a.u. (RMS)
* Info * 1.975266e-03 a.u. (Max)
* Info * Time : 4.60 sec
Optimization Step 117
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.540866636303 1.010410176504 0.651380759560
C -0.673882521861 0.359594496239 1.447421994899
O -0.237464696534 -0.906016900881 1.335872834596
H -1.801958279604 2.045936321145 0.875285214453
H -2.054484573267 0.519600879740 -0.178215922846
H -0.226265539184 0.857499155317 2.316231244837
H -0.398989439394 -1.247454429695 0.437288673443
C 0.296468832983 0.268429197455 -1.599602258495
O 0.019655051943 -0.896589097590 -1.447715411141
H 1.144046602600 0.759801903008 -1.065333254643
H -0.269558954023 0.926003678571 -2.303474914821
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000593093021 -0.000016888063 -0.000924967826
C 0.000821333934 -0.000001773297 -0.001036316909
O -0.000989621353 -0.000534323688 -0.000089087540
H 0.000152712949 0.000039106652 -0.000145691779
H -0.000117968038 -0.000074752589 0.000091579188
H 0.000189766750 0.000025838776 -0.000135550673
H -0.000090719007 -0.000161546822 -0.000067025290
C -0.001331611257 0.000329421578 0.001334734285
O 0.001223451502 0.000219971385 0.000851225797
H 0.000057581276 0.000076099833 -0.000289374578
H -0.000503639536 0.000102264669 0.000383672301
*** Time spent in gradient calculation: 1.77 sec ***
* Info * Energy : -268.1205678700 a.u.
* Info * Gradient : 9.924246e-04 a.u. (RMS)
* Info * 1.913955e-03 a.u. (Max)
* Info * Time : 4.46 sec
Optimization Step 118
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.547093560435 1.010725374275 0.660786956833
C -0.682403622519 0.359461465939 1.458397613791
O -0.230778334429 -0.900593165332 1.337709831423
H -1.821589923817 2.040615862067 0.894488267818
H -2.044555036510 0.525943872995 -0.182127935566
H -0.250010138521 0.851966875035 2.337930659165
H -0.381429231993 -1.234380630958 0.435757616983
C 0.311077994913 0.262046071269 -1.611736209476
O 0.011064896160 -0.896665309196 -1.456714921119
H 1.141737492344 0.747368999541 -1.046921879982
H -0.218234841101 0.918722058795 -2.344355663689
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000627113491 0.000017143056 -0.000773592924
C 0.000695907474 0.000071653211 -0.001099966296
O -0.001303851485 -0.001219854022 -0.001151163790
H 0.000114532892 0.000043157260 -0.000129426420
H -0.000091976311 -0.000075706913 0.000067042017
H 0.000233393023 0.000082954283 -0.000179775119
H 0.000183474210 0.000389086074 0.001229525410
C -0.001276590116 0.000432545557 0.000853228690
O 0.001405321158 0.000202845659 0.000870578251
H 0.000006765784 -0.000100012657 -0.000216483240
H -0.000589110098 0.000162538525 0.000508211519
*** Time spent in gradient calculation: 2.00 sec ***
* Info * Energy : -268.1208773178 a.u.
* Info * Gradient : 1.169806e-03 a.u. (RMS)
* Info * 2.124441e-03 a.u. (Max)
* Info * Time : 4.62 sec
Optimization Step 119
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.550968175699 1.010787266250 0.666620041887
C -0.687798362266 0.359113323052 1.465062198906
O -0.226325236897 -0.897411440074 1.338592415550
H -1.833555679698 2.037044039778 0.906683319128
H -2.037289018873 0.530944589411 -0.185589290087
H -0.264823711147 0.848447526254 2.350844911219
H -0.370239523901 -1.225389878666 0.434951390955
C 0.320157804388 0.258168290811 -1.619460101567
O 0.005158986384 -0.896430940093 -1.461640453420
H 1.135024571863 0.741917872964 -1.031294656238
H -0.181195012438 0.911495555822 -2.374185830557
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000653966478 -0.000028306131 -0.000617949535
C 0.000523857813 0.000248093089 -0.001181890238
O -0.001501125881 -0.002068878826 -0.002365867578
H 0.000101989912 0.000053058392 -0.000108967805
H -0.000030057542 -0.000052918860 0.000000810616
H 0.000272526810 0.000154800929 -0.000279709386
H 0.000415100172 0.000993935622 0.002641986780
C -0.001128701270 0.000995667744 0.000337971197
O 0.001389281003 -0.000268711413 0.001039316404
H -0.000049554492 -0.000226590984 -0.000112068635
H -0.000640477057 0.000207013164 0.000631824490
*** Time spent in gradient calculation: 1.95 sec ***
* Info * Energy : -268.1210803096 a.u.
* Info * Gradient : 1.634858e-03 a.u. (RMS)
* Info * 3.482954e-03 a.u. (Max)
* Info * Time : 4.17 sec
Optimization Step 120
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.553177489039 1.010931698566 0.670011149841
C -0.690854125165 0.358844011547 1.468955815827
O -0.223860679486 -0.895532539833 1.339210645288
H -1.840743438660 2.035036385105 0.913414879548
H -2.033323557281 0.533808701698 -0.187234602986
H -0.273131316130 0.846256207582 2.358345624358
H -0.364372225663 -1.220474926143 0.434524145348
C 0.325224266997 0.255733638672 -1.623906227593
O 0.002026123267 -0.896181554335 -1.464687257197
H 1.130996189224 0.738529390427 -1.022945112024
H -0.160818309206 0.907304371527 -2.390086967218
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000629877111 0.000031275092 -0.000582504867
C 0.000504775901 0.000161203199 -0.001220036876
O -0.001590007276 -0.002372948414 -0.002802935299
H 0.000061732196 0.000064151071 -0.000125361068
H -0.000007990393 -0.000033104609 -0.000028466519
H 0.000318530556 0.000209968028 -0.000286849486
H 0.000509795114 0.001240009161 0.003213220302
C -0.001106767830 0.000781868095 0.000093593890
O 0.001534254040 0.000033987115 0.001080342437
H -0.000133863869 -0.000360291094 -0.000042084095
H -0.000713449981 0.000251367165 0.000691260364
*** Time spent in gradient calculation: 2.04 sec ***
* Info * Energy : -268.1211940854 a.u.
* Info * Gradient : 1.845493e-03 a.u. (RMS)
* Info * 4.001931e-03 a.u. (Max)
* Info * Time : 4.14 sec
Optimization Step 121
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.555059398265 1.011049952217 0.672946056525
C -0.693407653547 0.358525482211 1.472293056942
O -0.221696731465 -0.893828488406 1.339669074060
H -1.846711482731 2.033402811988 0.918957726367
H -2.029442321723 0.536699786392 -0.189064864896
H -0.280055157412 0.844378897772 2.364725001649
H -0.360804560192 -1.216474173530 0.434150840867
C 0.329385268916 0.253540903348 -1.627621463298
O -0.000510746884 -0.895917303956 -1.467308667299
H 1.126593701321 0.735952464132 -1.015410791589
H -0.143193540160 0.903341008171 -2.403862251911
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000558057136 0.000071820671 -0.000549935186
C 0.000541988389 -0.000071259303 -0.001275196255
O -0.001624607780 -0.002458033982 -0.002910106447
H 0.000025189370 0.000085381183 -0.000157526618
H 0.000026562125 0.000005079294 -0.000080188150
H 0.000381883461 0.000289336304 -0.000254556146
H 0.000513794482 0.001376612429 0.003450549546
C -0.001135675718 0.000272193183 -0.000027178761
O 0.001759781334 0.000611280401 0.001087293756
H -0.000233175293 -0.000498471241 0.000013294406
H -0.000807651144 0.000324097440 0.000697450811
*** Time spent in gradient calculation: 2.09 sec ***
* Info * Energy : -268.1212927182 a.u.
* Info * Gradient : 1.946037e-03 a.u. (RMS)
* Info * 4.141256e-03 a.u. (Max)
* Info * Time : 4.57 sec
Optimization Step 122
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.557006479180 1.011126499618 0.675979278507
C -0.695959253760 0.357908052953 1.475844938007
O -0.219251520622 -0.891744138648 1.340052794577
H -1.852770780358 2.032005066595 0.923504114699
H -2.024374428996 0.540447615307 -0.191928403753
H -0.287057126955 0.842411166455 2.371536131937
H -0.360300517519 -1.212839055249 0.432634502999
C 0.333407918577 0.250920173341 -1.631064922705
O -0.002962622247 -0.895442478777 -1.470118633246
H 1.121536644418 0.733437613822 -1.007665481918
H -0.124846349913 0.898834983140 -2.418244531456
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000349301964 0.000133498138 -0.000510178905
C 0.000774962375 -0.000868460670 -0.001382028962
O -0.001432578202 -0.001703516656 -0.001435249718
H -0.000045860087 0.000152828288 -0.000282055946
H 0.000078735728 0.000107512113 -0.000186729838
H 0.000510312750 0.000453701150 -0.000092699740
H 0.000177082320 0.001021152975 0.002154028660
C -0.001417906909 -0.001387426892 0.000297600605
O 0.002328223069 0.002253919083 0.000898941315
H -0.000342929937 -0.000648603194 0.000008751245
H -0.000975348267 0.000494019278 0.000523952860
*** Time spent in gradient calculation: 1.91 sec ***
* Info * Energy : -268.1214015136 a.u.
* Info * Gradient : 1.767552e-03 a.u. (RMS)
* Info * 3.362866e-03 a.u. (Max)
* Info * Time : 4.48 sec
Optimization Step 123
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.559430389098 1.011129054851 0.679700677479
C -0.699346855894 0.357410615587 1.480301183316
O -0.216093252341 -0.889185668957 1.340299712247
H -1.859833063835 2.029974501797 0.930036483596
H -2.018917243122 0.544375174273 -0.194538867443
H -0.297159710162 0.838899562446 2.380720140367
H -0.356203263403 -1.207969056848 0.430460697350
C 0.338882168643 0.248061364555 -1.635293812402
O -0.006670800632 -0.895126645601 -1.473504785879
H 1.115014840322 0.731750148829 -0.998035065279
H -0.099065314111 0.891846436853 -2.437566699501
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000194596170 0.000094757943 -0.000490576629
C 0.000926737222 -0.001068819805 -0.001341598466
O -0.001273602835 -0.001209238182 -0.000274856994
H -0.000027732906 0.000182274513 -0.000356703297
H 0.000104647444 0.000163121527 -0.000247185638
H 0.000532992212 0.000466758195 -0.000056188436
H -0.000054520900 0.000655409401 0.001099993527
C -0.001537635953 -0.001862951631 0.000531933468
O 0.002492759843 0.002677166651 0.000723768723
H -0.000318827696 -0.000633865161 -0.000021012419
H -0.001037699526 0.000543955853 0.000427704751
*** Time spent in gradient calculation: 2.04 sec ***
* Info * Energy : -268.1215486951 a.u.
* Info * Gradient : 1.698617e-03 a.u. (RMS)
* Info * 3.728929e-03 a.u. (Max)
* Info * Time : 4.53 sec
Optimization Step 124
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.562365582024 1.011077146087 0.684466242696
C -0.704051332533 0.357003691902 1.486262951067
O -0.211186556315 -0.885316118121 1.338955976521
H -1.868875818576 2.026688685177 0.939693660040
H -2.014216788154 0.547103810940 -0.195129538325
H -0.314623227762 0.829947912984 2.395827071388
H -0.348404280802 -1.203159010732 0.422879300140
C 0.346854895742 0.245108551571 -1.639842249861
O -0.013205257446 -0.895356726338 -1.476663897214
H 1.105060664072 0.736610662164 -0.985770336770
H -0.053490995126 0.875564944984 -2.470838472247
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000250075706 -0.000124531101 -0.000609456320
C 0.001104044549 0.000059696677 -0.000790683853
O -0.000678093733 0.000293572197 0.003432329860
H 0.000201117497 0.000113811109 -0.000388450354
H -0.000024144350 0.000050365754 -0.000103121323
H 0.000181711798 -0.000017680006 -0.000259693994
H -0.000673966806 -0.001169331410 -0.002872856581
C -0.001595340652 0.000754706522 0.001259721493
O 0.001429460444 -0.000400003413 0.000246558660
H 0.000453641001 0.000236854918 -0.000301013523
H -0.000646700367 0.000207278120 0.000376457913
*** Time spent in gradient calculation: 2.08 sec ***
* Info * Energy : -268.1217849839 a.u.
* Info * Gradient : 1.730446e-03 a.u. (RMS)
* Info * 3.510966e-03 a.u. (Max)
* Info * Time : 4.95 sec
Optimization Step 125
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.564364946814 1.011193799160 0.687708143678
C -0.707145704518 0.356179120822 1.490138404242
O -0.208042287893 -0.882782366086 1.338281831883
H -1.875766796964 2.024427113050 0.946791322115
H -2.008973193482 0.551397199849 -0.197781736831
H -0.323463071523 0.827024015958 2.403937275841
H -0.343260533467 -1.196272494757 0.421957094860
C 0.351795966511 0.241694669484 -1.643470578790
O -0.016843236691 -0.894436326454 -1.478903773912
H 1.094146017297 0.735043272988 -0.973985299419
H -0.026462276825 0.867031452643 -2.489456165283
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000126070153 -0.000115813562 -0.000750847221
C 0.001201067046 -0.000649390741 -0.001003469490
O -0.000979164076 0.000020168079 0.002262106681
H 0.000185362352 0.000176509294 -0.000393444156
H 0.000008753344 0.000087261875 -0.000166653035
H 0.000358748903 0.000232198240 0.000014286906
H -0.000489025148 -0.000550238113 -0.001551657845
C -0.001834584957 -0.001142754643 0.001710714782
O 0.002148262433 0.001658651885 0.000080835703
H 0.000132561376 -0.000113252192 -0.000369652711
H -0.000854321178 0.000402749569 0.000160042966
*** Time spent in gradient calculation: 1.92 sec ***
* Info * Energy : -268.1219407699 a.u.
* Info * Gradient : 1.623249e-03 a.u. (RMS)
* Info * 2.756472e-03 a.u. (Max)
* Info * Time : 5.60 sec
Optimization Step 126
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.567212028063 1.011515195828 0.692307191041
C -0.711468707837 0.355345785023 1.494952210177
O -0.203581446867 -0.879496098873 1.336839153736
H -1.887176782310 2.020167018096 0.958798827882
H -2.004560792869 0.555594043934 -0.198722548222
H -0.336188127030 0.821937058489 2.414567216917
H -0.334531638992 -1.187773592470 0.420612107419
C 0.359577527067 0.237596313238 -1.648861182305
O -0.022454914218 -0.893521996465 -1.481240011656
H 1.078958758394 0.734456219639 -0.958163364974
H 0.014260836131 0.853571527373 -2.515447047054
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000352342093 -0.000076423582 -0.000898034656
C 0.001036915149 -0.000670321836 -0.001189864965
O -0.001357839858 -0.000822928677 0.000173161725
H 0.000154900612 0.000166464378 -0.000299399447
H -0.000011033962 0.000023803372 -0.000090087653
H 0.000388615444 0.000324132175 0.000075582278
H -0.000089649278 0.000223117008 0.000648948697
C -0.001704113019 -0.001133184001 0.001415280104
O 0.002274551379 0.001916425300 0.000250365961
H -0.000089510235 -0.000397112911 -0.000290552800
H -0.000946319999 0.000452373944 0.000201722960
*** Time spent in gradient calculation: 1.94 sec ***
* Info * Energy : -268.1221655705 a.u.
* Info * Gradient : 1.471228e-03 a.u. (RMS)
* Info * 2.984787e-03 a.u. (Max)
* Info * Time : 4.21 sec
Optimization Step 127
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.570704285144 1.011958071960 0.698146970849
C -0.717021413316 0.354393138569 1.501052352864
O -0.197984216419 -0.875347326226 1.334549193306
H -1.901425591392 2.014370829432 0.974480888170
H -1.999607841036 0.560332285238 -0.199031282448
H -0.353082541084 0.813900335353 2.428245971876
H -0.323582774423 -1.178170731281 0.416898030047
C 0.369288239942 0.232979280339 -1.655203890831
O -0.029674297637 -0.892904779699 -1.484030577486
H 1.059907410640 0.737586646954 -0.941165007878
H 0.068411233914 0.833773813932 -2.547577338956
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000728319572 -0.000083963381 -0.001093243313
C 0.000758609213 0.000040451826 -0.001033069712
O -0.001556385269 -0.001484597322 -0.001122024487
H 0.000193076507 0.000072465238 -0.000143321282
H -0.000064599735 -0.000113513040 0.000057391844
H 0.000258753827 0.000197118218 -0.000125876252
H 0.000230728908 0.000487934458 0.001905879534
C -0.001459954063 0.000611623295 0.000867488349
O 0.001671644615 0.000250924276 0.000428405332
H 0.000049902462 -0.000277528858 -0.000192309186
H -0.000799096313 0.000307764225 0.000454250423
*** Time spent in gradient calculation: 1.76 sec ***
* Info * Energy : -268.1224538385 a.u.
* Info * Gradient : 1.373324e-03 a.u. (RMS)
* Info * 2.425964e-03 a.u. (Max)
* Info * Time : 3.94 sec
Optimization Step 128
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.573684219591 1.012386357645 0.703164988647
C -0.721315356645 0.353638925518 1.505863791390
O -0.193470585501 -0.871948069769 1.332431185705
H -1.912746089070 2.009901563848 0.986590921562
H -1.995427134564 0.564786005119 -0.199386155622
H -0.366470261927 0.806554332751 2.439073893314
H -0.317233890261 -1.171564183887 0.411737693331
C 0.376860911949 0.229459627864 -1.660012023069
O -0.035411672376 -0.892638380454 -1.485972233611
H 1.044942372720 0.742426000323 -0.929900943544
H 0.112693279912 0.816115246309 -2.572161418799
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.001011345849 -0.000136103629 -0.001104768693
C 0.000582485212 0.000828894409 -0.000758926334
O -0.001340589958 -0.001328946859 -0.000056521330
H 0.000227765106 -0.000013865759 -0.000087316961
H -0.000115323084 -0.000198212041 0.000119725613
H 0.000089124456 -0.000028051046 -0.000424529629
H 0.000134630327 -0.000060482132 0.000772753340
C -0.001281722788 0.002564154646 0.000360242691
O 0.000867409403 -0.001864027926 0.000541758553
H 0.000422008573 0.000099514455 -0.000062771679
H -0.000590001809 0.000149062294 0.000704674669
*** Time spent in gradient calculation: 1.91 sec ***
* Info * Energy : -268.1226858847 a.u.
* Info * Gradient : 1.423567e-03 a.u. (RMS)
* Info * 2.889200e-03 a.u. (Max)
* Info * Time : 4.20 sec
Optimization Step 129
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.577908500194 1.012915009743 0.709983616646
C -0.726709945981 0.352105433960 1.512227053527
O -0.187591455886 -0.867212001318 1.329649045127
H -1.926815173587 2.004784101888 1.001217708483
H -1.988597631865 0.572553384550 -0.201223195532
H -0.382751223491 0.797801459408 2.453169744289
H -0.309637460683 -1.160806342212 0.405944780406
C 0.386327788062 0.223699036791 -1.666096778992
O -0.042618183433 -0.891323044211 -1.488696996785
H 1.022043355771 0.747508738561 -0.915560408912
H 0.169738599087 0.792054888318 -2.602315342981
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000996537760 -0.000111909572 -0.001217499984
C 0.000670682482 0.000613762452 -0.000807226832
O -0.001340117426 -0.001148160173 0.000435584277
H 0.000214237948 0.000002325655 -0.000105552872
H -0.000130192731 -0.000201030005 0.000089664604
H 0.000129570158 0.000032438922 -0.000324469531
H 0.000145263797 -0.000158781020 0.000393205172
C -0.001370913841 0.002125644943 0.000722731987
O 0.000929988284 -0.001428813583 0.000377569271
H 0.000379205000 0.000063764620 -0.000096265549
H -0.000623948152 0.000226433246 0.000535981377
*** Time spent in gradient calculation: 1.75 sec ***
* Info * Energy : -268.1229906396 a.u.
* Info * Gradient : 1.299318e-03 a.u. (RMS)
* Info * 2.630611e-03 a.u. (Max)
* Info * Time : 4.29 sec
Optimization Step 130
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.585076132930 1.013824641385 0.719738820448
C -0.732166034814 0.349550497651 1.518987527020
O -0.180523923874 -0.861181652787 1.324157543596
H -1.943970125531 2.000572194708 1.017059237626
H -1.978669930210 0.587547322272 -0.205921487923
H -0.399267248428 0.786686785633 2.469563923551
H -0.312946318274 -1.147982543448 0.395385901402
C 0.398231905086 0.214455119046 -1.673732306413
O -0.050018688192 -0.888253177075 -1.489386205488
H 0.998173643976 0.752194154708 -0.905149523828
H 0.233267447648 0.760821586659 -2.635977928193
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000109459085 0.000031542661 -0.001230807838
C 0.001623721533 -0.001248063140 -0.001488653784
O -0.000690862394 0.000232607309 0.004100220806
H 0.000050055493 0.000247739436 -0.000460543066
H -0.000051471166 0.000088264913 -0.000239474522
H 0.000429753437 0.000415648919 0.000481346719
H -0.000779002236 -0.000773686101 -0.002836637743
C -0.002603824487 -0.002975226743 0.002739510109
O 0.002900254234 0.003570965757 -0.000208026306
H -0.000046507330 -0.000365804779 -0.000343205152
H -0.000947147276 0.000788610380 -0.000519693109
*** Time spent in gradient calculation: 1.81 sec ***
* Info * Energy : -268.1233227262 a.u.
* Info * Gradient : 2.727842e-03 a.u. (RMS)
* Info * 4.810072e-03 a.u. (Max)
* Info * Time : 4.33 sec
Optimization Step 131
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.586680645884 1.014273408335 0.723407730528
C -0.735639941526 0.349306458228 1.522476609502
O -0.177535456104 -0.858571982043 1.322315762415
H -1.951526099542 1.995639050212 1.030082406970
H -1.978368690649 0.588652518234 -0.203097261772
H -0.408656792790 0.781052858547 2.475874184924
H -0.300481112436 -1.142704029800 0.395221182852
C 0.404294125046 0.212663488279 -1.677417332040
O -0.054486888662 -0.888830161506 -1.491142940593
H 0.984381346932 0.758520985871 -0.898627417553
H 0.269201060260 0.744223550874 -2.649659043469
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000740340190 -0.000063878489 -0.001428174167
C 0.000941104861 0.000398652074 -0.000639131425
O -0.001415066174 -0.001003307455 0.000461166293
H 0.000255216578 -0.000019813423 -0.000083778210
H -0.000092233274 -0.000186496066 0.000004092493
H 0.000144368465 0.000044913882 -0.000327548609
H 0.000242535228 -0.000203502615 0.000402014040
C -0.001248767824 0.001954139768 0.000815682032
O 0.000805616440 -0.001107714294 0.000370129233
H 0.000287002706 -0.000002080721 -0.000045627940
H -0.000665480961 0.000196236442 0.000472893921
*** Time spent in gradient calculation: 1.84 sec ***
* Info * Energy : -268.1235210732 a.u.
* Info * Gradient : 1.229207e-03 a.u. (RMS)
* Info * 2.458337e-03 a.u. (Max)
* Info * Time : 4.46 sec
Optimization Step 132
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.592284659949 1.015255994095 0.732749735582
C -0.741770486022 0.347134350170 1.529647326039
O -0.170853233708 -0.852650190209 1.318162210567
H -1.969170818580 1.989871614779 1.046897547377
H -1.971359179678 0.599663384965 -0.203609034175
H -0.426286310656 0.769734652654 2.491859795898
H -0.297121896348 -1.129136680929 0.387038560325
C 0.415652006521 0.204621237280 -1.684858449450
O -0.061944312101 -0.886627987925 -1.494044716292
H 0.957955338187 0.766396138457 -0.890956750399
H 0.335302600779 0.711779503946 -2.677231460847
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000844792188 -0.000105150259 -0.001201223797
C 0.000902124633 -0.000246468734 -0.001370842187
O -0.001029949051 -0.000581300596 0.002237412423
H 0.000087882680 0.000124242251 -0.000266947105
H -0.000125935525 -0.000092103428 -0.000033935033
H 0.000264278992 0.000257089176 0.000106557221
H -0.000042230942 -0.000391032123 -0.001084241184
C -0.001829213939 -0.000164215062 0.001782906697
O 0.001518784569 0.000878787222 0.000073251756
H 0.000091603161 -0.000180137309 -0.000157567913
H -0.000693501153 0.000504170054 -0.000086248162
*** Time spent in gradient calculation: 1.78 sec ***
* Info * Energy : -268.1238576036 a.u.
* Info * Gradient : 1.457519e-03 a.u. (RMS)
* Info * 2.559638e-03 a.u. (Max)
* Info * Time : 4.07 sec
Optimization Step 133
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.596809738078 1.016030724890 0.739894422780
C -0.746471194405 0.345929119118 1.535400511410
O -0.166134399717 -0.847993390501 1.314526792188
H -1.981405394809 1.984219099543 1.063674660592
H -1.967151912162 0.607114467303 -0.202836180454
H -0.439482150581 0.759986917127 2.503081761269
H -0.293056746748 -1.121108629104 0.383840056418
C 0.425155184539 0.199324015132 -1.690862954281
O -0.067632525827 -0.885889576604 -1.496341816870
H 0.939368623929 0.774303649149 -0.887337696623
H 0.386464876484 0.684687215833 -2.695141168009
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000599067339 0.000059537269 -0.001609291992
C 0.001054976007 0.000282859415 -0.000481266844
O -0.001240510168 -0.000962920604 0.000787559826
H 0.000252258033 -0.000032984634 -0.000093377424
H -0.000102323131 -0.000197287950 -0.000021581230
H 0.000105578467 0.000022537695 -0.000335457018
H 0.000304204884 -0.000232518608 0.000159575368
C -0.001132917266 0.001778252781 0.000823761096
O 0.000542447592 -0.000906041461 0.000315668002
H 0.000220288369 0.000003230618 0.000046226548
H -0.000613307577 0.000187871645 0.000407325739
*** Time spent in gradient calculation: 1.85 sec ***
* Info * Energy : -268.1241086530 a.u.
* Info * Gradient : 1.161889e-03 a.u. (RMS)
* Info * 2.263684e-03 a.u. (Max)
* Info * Time : 4.18 sec
Optimization Step 134
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.603695186823 1.017483735251 0.751390419526
C -0.752972744913 0.343964308013 1.542746643776
O -0.159779855107 -0.841267002464 1.308001747479
H -2.000803863466 1.977988219456 1.083444712773
H -1.960527863825 0.620231288962 -0.201441124902
H -0.455709916925 0.746861111131 2.518657548407
H -0.301528691873 -1.109442215197 0.377702317927
C 0.437680697588 0.190271139970 -1.699587474181
O -0.073732306713 -0.883900515601 -1.497988368779
H 0.922249765887 0.781188250934 -0.889134120307
H 0.445933598068 0.651019797873 -2.716833174142
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000887579302 -0.000249577615 -0.001148961976
C 0.000583272608 0.000167211319 -0.001314285151
O -0.000501630607 -0.001138475699 0.001063851978
H 0.000078940695 0.000124575788 -0.000268943928
H -0.000032414059 -0.000062591992 0.000016032967
H 0.000143778321 0.000133116865 -0.000021347959
H -0.000264281253 -0.000049592180 -0.000003728061
C -0.001603863365 0.000204784881 0.001048817911
O 0.001128055904 0.000449772445 0.000461963819
H 0.000135840045 -0.000046924707 0.000066256999
H -0.000561011898 0.000470693830 0.000100124174
*** Time spent in gradient calculation: 1.77 sec ***
* Info * Energy : -268.1244213607 a.u.
* Info * Gradient : 1.092521e-03 a.u. (RMS)
* Info * 1.927261e-03 a.u. (Max)
* Info * Time : 4.55 sec
Optimization Step 135
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.610832980724 1.018890348780 0.763254815545
C -0.759446265134 0.342073528571 1.551270577745
O -0.154779569352 -0.834219518426 1.302372668689
H -2.017399891686 1.970952662798 1.107279718744
H -1.957197719997 0.631488043856 -0.197652076923
H -0.471051684098 0.734082138608 2.533967730437
H -0.292257810254 -1.094651124508 0.368690626636
C 0.450651432961 0.181882463972 -1.708561400119
O -0.079727205334 -0.882907764314 -1.501579920546
H 0.904112361317 0.790318051993 -0.893419648598
H 0.508106475035 0.612160573161 -2.736821059153
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000631056097 -0.000116422028 -0.001447790031
C 0.000754427920 0.000177641132 -0.000428640762
O -0.000931548375 -0.000730828304 0.000919893553
H 0.000244625397 -0.000024153556 -0.000105658187
H -0.000047990721 -0.000177572151 -0.000072365358
H 0.000143408437 0.000143940507 -0.000185290922
H 0.000182673188 -0.000282533410 -0.000255525305
C -0.001006268558 0.001493352867 0.001068793905
O 0.000404987429 -0.000551204286 0.000333809268
H 0.000137925223 -0.000089263220 -0.000025156673
H -0.000516257441 0.000156986040 0.000201108758
*** Time spent in gradient calculation: 1.76 sec ***
* Info * Energy : -268.1247182409 a.u.
* Info * Gradient : 1.010342e-03 a.u. (RMS)
* Info * 2.094039e-03 a.u. (Max)
* Info * Time : 4.30 sec
Optimization Step 136
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.618202858929 1.020903941632 0.775826574323
C -0.766056998478 0.340128896945 1.559036315335
O -0.149280251169 -0.827283418647 1.296692258608
H -2.036784665195 1.964568895325 1.128938470154
H -1.953092602809 0.645339643722 -0.193756321945
H -0.486811297236 0.719701740665 2.549147660315
H -0.298233944227 -1.082770609260 0.364333523150
C 0.463488189199 0.172552091665 -1.718094273328
O -0.084759025568 -0.881379136663 -1.504896215918
H 0.890228732609 0.797174229262 -0.900161930676
H 0.563772848959 0.578075245133 -2.753250111586
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000803507908 -0.000227425167 -0.001123120540
C 0.000461405745 0.000164938957 -0.000888771732
O -0.000430725653 -0.001125856292 0.000304534194
H 0.000075163642 0.000101549438 -0.000241713063
H -0.000033690550 -0.000069348926 0.000020814997
H 0.000097777195 0.000060270881 -0.000167259486
H -0.000073040061 0.000108925930 0.000547242135
C -0.001130407118 0.000584081837 0.000685788336
O 0.000584305673 0.000087489141 0.000550106839
H 0.000086384731 -0.000014810602 0.000158057768
H -0.000447485061 0.000334043513 0.000164218423
*** Time spent in gradient calculation: 1.79 sec ***
* Info * Energy : -268.1249801871 a.u.
* Info * Gradient : 8.573537e-04 a.u. (RMS)
* Info * 1.445433e-03 a.u. (Max)
* Info * Time : 4.20 sec
Optimization Step 137
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.625623186414 1.022553959994 0.788281175648
C -0.771941544732 0.338436007263 1.566968535355
O -0.145209189177 -0.820404362859 1.291237443313
H -2.052448416426 1.958146310776 1.152390808941
H -1.950611461367 0.657833759205 -0.188998169445
H -0.500455397826 0.706822245369 2.563645303114
H -0.294241108977 -1.069719585405 0.355659865620
C 0.475496895739 0.164045555726 -1.726967320693
O -0.089071535427 -0.880472829232 -1.508727727543
H 0.878703683864 0.804750841887 -0.909802076117
H 0.616056186809 0.542145838891 -2.767756308147
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000530747203 -0.000192618957 -0.001210211256
C 0.000578920439 0.000166725424 -0.000524558761
O -0.000507490115 -0.000581415737 0.001161640386
H 0.000205119775 0.000008819183 -0.000126563132
H -0.000018669630 -0.000130868326 -0.000087097045
H 0.000115595962 0.000133803987 -0.000041973372
H 0.000015392496 -0.000324344389 -0.000616737358
C -0.000938845862 0.001004714633 0.001025410446
O 0.000274814819 -0.000216134007 0.000368497597
H 0.000090007702 -0.000049526336 -0.000004799624
H -0.000354292535 0.000184290221 0.000062207912
*** Time spent in gradient calculation: 1.85 sec ***
* Info * Energy : -268.1252037903 a.u.
* Info * Gradient : 8.708869e-04 a.u. (RMS)
* Info * 1.715328e-03 a.u. (Max)
* Info * Time : 4.24 sec
Optimization Step 138
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.632326966000 1.024738188831 0.800208126944
C -0.777674373170 0.336805210026 1.574095624031
O -0.141468138393 -0.814082949322 1.286355797124
H -2.069643806937 1.952088932030 1.173066585100
H -1.948773385395 0.669981999877 -0.183496010061
H -0.512744836756 0.694283939239 2.576122147821
H -0.297557385636 -1.058732491322 0.352459710245
C 0.486616839113 0.155799424754 -1.735738043772
O -0.092155305839 -0.879724887692 -1.512497037989
H 0.871535516631 0.809180255204 -0.919295547328
H 0.658138907647 0.513139608988 -2.778933010795
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000660714346 -0.000135391203 -0.001018033775
C 0.000443442127 0.000086601165 -0.000597877027
O -0.000418847507 -0.000972763575 -0.000178335072
H 0.000055156811 0.000051601466 -0.000185391131
H -0.000007573334 -0.000088483999 0.000010114012
H 0.000054499176 0.000043697713 -0.000217362162
H 0.000066172596 0.000148725343 0.000816880974
C -0.000771708421 0.000738349788 0.000504498547
O 0.000223568468 -0.000057553590 0.000522790972
H 0.000027407952 -0.000004337496 0.000154755325
H -0.000339681804 0.000191576214 0.000194854349
*** Time spent in gradient calculation: 1.96 sec ***
* Info * Energy : -268.1253827335 a.u.
* Info * Gradient : 7.346579e-04 a.u. (RMS)
* Info * 1.221174e-03 a.u. (Max)
* Info * Time : 4.33 sec
Optimization Step 139
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.638720005674 1.026290616307 0.811131356471
C -0.782480678423 0.335512566449 1.580601726272
O -0.138594251739 -0.808338636625 1.282221986283
H -2.081711729173 1.947385391008 1.192600727922
H -1.947915865794 0.681513164926 -0.178586899747
H -0.523213457891 0.683425965440 2.587908409406
H -0.296495694848 -1.048264340509 0.345428790043
C 0.496146176086 0.148544238110 -1.743394009147
O -0.094544654565 -0.879198111008 -1.516286453983
H 0.867082274964 0.813159530870 -0.929658069887
H 0.694435335553 0.486576253437 -2.788695788101
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000510609238 -0.000261141880 -0.000985254229
C 0.000407379001 0.000134472401 -0.000627725157
O -0.000221154918 -0.000458147125 0.001231956424
H 0.000144824307 0.000039843171 -0.000120042117
H -0.000015975768 -0.000080186811 -0.000075895646
H 0.000098706681 0.000111900661 0.000055174324
H -0.000089688363 -0.000288141500 -0.000740512555
C -0.000910487837 0.000485617810 0.000933658927
O 0.000262298501 0.000138898945 0.000350232743
H 0.000022621696 -0.000029613021 0.000032031559
H -0.000212812850 0.000203941260 -0.000051207776
*** Time spent in gradient calculation: 1.78 sec ***
* Info * Energy : -268.1255234406 a.u.
* Info * Gradient : 7.750430e-04 a.u. (RMS)
* Info * 1.391593e-03 a.u. (Max)
* Info * Time : 4.14 sec
Optimization Step 140
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.644740557240 1.028416410386 0.821853597019
C -0.787230498910 0.334264102949 1.586863573729
O -0.136206454652 -0.802825063637 1.277946734866
H -2.095844579332 1.942119898487 1.211347959539
H -1.947576672844 0.691672773512 -0.172606450614
H -0.532344289367 0.672961384848 2.597855225151
H -0.298217919644 -1.038941504297 0.342700774474
C 0.505517522599 0.141627622636 -1.751174830504
O -0.096199841530 -0.879092870894 -1.519745739251
H 0.866586752239 0.815113692544 -0.939920992342
H 0.723961219095 0.463232724304 -2.796980732541
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000449370741 -0.000018065852 -0.000975497405
C 0.000452354041 0.000065698661 -0.000259707718
O -0.000355684318 -0.000771113075 -0.000283962576
H 0.000081691630 -0.000010006130 -0.000116854356
H 0.000012330781 -0.000106522348 -0.000016339988
H 0.000022960379 0.000021498474 -0.000246260665
H 0.000109965973 0.000072284650 0.000714832192
C -0.000487655300 0.000935153515 0.000336809022
O -0.000059762597 -0.000303933566 0.000479492751
H 0.000020778979 0.000031346365 0.000132621752
H -0.000247261726 0.000078224232 0.000239743366
*** Time spent in gradient calculation: 1.75 sec ***
* Info * Energy : -268.1256417988 a.u.
* Info * Gradient : 6.431099e-04 a.u. (RMS)
* Info * 1.107141e-03 a.u. (Max)
* Info * Time : 3.97 sec
Optimization Step 141
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.650757605683 1.030008716206 0.832629955087
C -0.791791035965 0.333123199624 1.592942181537
O -0.133953063790 -0.797402155962 1.274481021621
H -2.107436714281 1.938009516168 1.229182150668
H -1.947620387898 0.702811167733 -0.166922561548
H -0.541472864102 0.663011459340 2.608588803691
H -0.298153197142 -1.028563959464 0.336856371730
C 0.514204693739 0.134587340261 -1.758739292633
O -0.097527765891 -0.878717188258 -1.523865872281
H 0.866697989061 0.816052386415 -0.950501901827
H 0.751567056990 0.441791466783 -2.805479473538
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000580997320 -0.000304722110 -0.000712777794
C 0.000203914648 0.000086858268 -0.000744610080
O -0.000096988315 -0.000437775849 0.000948580217
H 0.000060262127 0.000066285851 -0.000120998233
H -0.000010062425 -0.000046568781 -0.000042033211
H 0.000092063503 0.000116615109 0.000111454994
H -0.000089927841 -0.000148435728 -0.000506839689
C -0.000868694444 0.000093974760 0.000873207282
O 0.000307232429 0.000423494785 0.000277847537
H -0.000057908156 -0.000046882880 0.000036555838
H -0.000119104064 0.000189030425 -0.000118239603
*** Time spent in gradient calculation: 1.78 sec ***
* Info * Energy : -268.1257434922 a.u.
* Info * Gradient : 6.700596e-04 a.u. (RMS)
* Info * 1.235294e-03 a.u. (Max)
* Info * Time : 4.33 sec
Optimization Step 142
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.657500412353 1.032223989862 0.843950349064
C -0.796427133908 0.332034054465 1.599359904095
O -0.132099433746 -0.791675136032 1.270144836857
H -2.120146171003 1.933027442025 1.249434085290
H -1.948737966822 0.713681561758 -0.160140055869
H -0.549851817138 0.652200168869 2.618345404629
H -0.299867469833 -1.020017769011 0.333470903690
C 0.523836088335 0.127536832949 -1.767016032646
O -0.098490657776 -0.878935385085 -1.527413023526
H 0.873133438581 0.815463701988 -0.962373307479
H 0.775090953409 0.420260515214 -2.813834686926
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000182851827 0.000069379794 -0.000953202186
C 0.000500743211 0.000077020094 0.000063988053
O -0.000224948858 -0.000502445629 -0.000038045922
H 0.000135476210 -0.000048322281 -0.000054647968
H 0.000018608044 -0.000103503054 -0.000057023030
H -0.000004081504 -0.000028396929 -0.000240151448
H 0.000049159747 -0.000088342884 0.000298445710
C -0.000278483312 0.000995788819 0.000166621397
O -0.000246303460 -0.000504269000 0.000438528355
H 0.000036712376 0.000097737687 0.000130470574
H -0.000165094114 0.000026122271 0.000246728036
*** Time spent in gradient calculation: 1.73 sec ***
* Info * Energy : -268.1258367580 a.u.
* Info * Gradient : 5.583086e-04 a.u. (RMS)
* Info * 1.047335e-03 a.u. (Max)
* Info * Time : 3.93 sec
Optimization Step 143
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.663965567554 1.034100304680 0.856227246694
C -0.801489897601 0.330842443627 1.606035733210
O -0.130138032850 -0.785693859778 1.266753262539
H -2.134105795009 1.928206038534 1.268371831159
H -1.949528442644 0.725176713750 -0.152533038727
H -0.558889194196 0.642199348095 2.629349598557
H -0.299135027998 -1.007402984335 0.328359771746
C 0.533201272962 0.119700960967 -1.775743703287
O -0.099226123250 -0.878613830168 -1.532470826260
H 0.877889321529 0.813404223280 -0.974382873712
H 0.798903888188 0.399917332828 -2.823165815337
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000660682878 -0.000285303805 -0.000407628779
C 0.000023462057 0.000010987006 -0.000812379628
O -0.000111898305 -0.000477493692 0.000380702590
H -0.000031791114 0.000073559176 -0.000127560468
H 0.000003691598 -0.000026469451 0.000005743926
H 0.000078321603 0.000139049897 0.000113184964
H 0.000007981494 0.000053180127 0.000004038644
C -0.000765156529 -0.000104146193 0.000777099045
O 0.000336434763 0.000543840643 0.000164433700
H -0.000127283334 -0.000075011134 0.000011514036
H -0.000065510670 0.000137627990 -0.000111701688
*** Time spent in gradient calculation: 1.95 sec ***
* Info * Energy : -268.1259213034 a.u.
* Info * Gradient : 5.631593e-04 a.u. (RMS)
* Info * 1.095534e-03 a.u. (Max)
* Info * Time : 4.25 sec
Optimization Step 144
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.672021462215 1.036305034177 0.868859110022
C -0.806278042434 0.329899914369 1.612757809758
O -0.128559958254 -0.779307425958 1.261992757890
H -2.145912014425 1.923403588144 1.291214049573
H -1.952573753532 0.737772159844 -0.144609816910
H -0.566955882411 0.630255476447 2.639609582538
H -0.302043987963 -0.999205531845 0.323521366392
C 0.543828591301 0.111702168430 -1.785068889249
O -0.099698157251 -0.878795436584 -1.535925872017
H 0.893215130591 0.808797906579 -0.988032450126
H 0.816829694984 0.379794681869 -2.833234604966
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000026548776 0.000033995167 -0.000826985958
C 0.000443578991 0.000122676555 0.000144908724
O -0.000043888977 -0.000194841262 0.000512702695
H 0.000169857758 -0.000040622329 -0.000002798682
H 0.000016309459 -0.000075879945 -0.000096842562
H 0.000000499832 -0.000059605255 -0.000118858189
H -0.000101933783 -0.000275650990 -0.000379376289
C -0.000277583665 0.000716318318 0.000151363534
O -0.000191455865 -0.000419421494 0.000363699723
H 0.000041820376 0.000137286929 0.000116753684
H -0.000069435777 0.000045434355 0.000125326687
*** Time spent in gradient calculation: 1.82 sec ***
* Info * Energy : -268.1259973318 a.u.
* Info * Gradient : 4.795799e-04 a.u. (RMS)
* Info * 8.281101e-04 a.u. (Max)
* Info * Time : 4.49 sec
Optimization Step 145
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.677280573683 1.037896178673 0.878899141358
C -0.810313063394 0.329035342349 1.618099564431
O -0.127233816078 -0.774390674263 1.259561775333
H -2.158172968836 1.919021737307 1.306142112674
H -1.954124677738 0.746065164282 -0.137507092029
H -0.573358289428 0.623262527137 2.647515042518
H -0.300250080525 -0.988275919100 0.320672977702
C 0.551530948315 0.105285138977 -1.792398757484
O -0.100046376643 -0.878594026650 -1.540249204765
H 0.901345326563 0.804465427276 -0.997745316543
H 0.831472065553 0.365278600603 -2.840965940078
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000482492348 -0.000090516876 -0.000341791292
C 0.000086670066 -0.000061006482 -0.000477728774
O -0.000190390381 -0.000465857455 -0.000188559181
H -0.000035429500 0.000028117329 -0.000099513726
H 0.000016809011 -0.000030240184 0.000018126655
H 0.000033716615 0.000099208808 -0.000019673652
H 0.000108115714 0.000135221112 0.000455670562
C -0.000465564822 0.000168635927 0.000441050931
O 0.000165724583 0.000194289216 0.000154563259
H -0.000115851954 -0.000042219357 0.000016292540
H -0.000067804541 0.000054218699 0.000027420602
*** Time spent in gradient calculation: 1.80 sec ***
* Info * Energy : -268.1260494354 a.u.
* Info * Gradient : 3.932526e-04 a.u. (RMS)
* Info * 6.631098e-04 a.u. (Max)
* Info * Time : 4.15 sec
Optimization Step 146
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.683484318313 1.039183773201 0.888565798011
C -0.813874924029 0.328495136207 1.623131313171
O -0.125967197573 -0.769350564707 1.256457709906
H -2.166998847326 1.915358910833 1.322820404828
H -1.956826855548 0.755345393113 -0.131191251374
H -0.579191482802 0.614636900094 2.655296903231
H -0.302080599545 -0.981521859146 0.316722123079
C 0.559628888055 0.098852310649 -1.799607958148
O -0.100459468697 -0.878402697346 -1.543275075474
H 0.915828644153 0.798570311538 -1.007870416474
H 0.842488729664 0.351806307310 -2.849156213174
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000186743494 -0.000061960562 -0.000500689692
C 0.000217277218 0.000096473124 -0.000150703569
O -0.000015720741 -0.000179288898 0.000545875187
H 0.000086531131 0.000002751804 -0.000025697050
H 0.000013309083 -0.000035773692 -0.000064014415
H 0.000023612874 -0.000009555575 -0.000001125487
H -0.000089562922 -0.000174215406 -0.000387395239
C -0.000411245810 0.000273114190 0.000289410749
O 0.000055015252 -0.000068174526 0.000208100594
H -0.000026066311 0.000071752971 0.000066480743
H -0.000018000977 0.000074886647 -0.000001419384
*** Time spent in gradient calculation: 1.77 sec ***
* Info * Energy : -268.1260952754 a.u.
* Info * Gradient : 3.433054e-04 a.u. (RMS)
* Info * 5.747794e-04 a.u. (Max)
* Info * Time : 3.92 sec
Optimization Step 147
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.689359059703 1.040543548743 0.898455060490
C -0.817574320762 0.327850069517 1.628179371435
O -0.124726127288 -0.764223361962 1.253608343089
H -2.178203272179 1.910914535904 1.338425728272
H -1.959698648711 0.763354158709 -0.123939623474
H -0.584576316176 0.607699588257 2.662413983725
H -0.301701036078 -0.972263101303 0.313963386562
C 0.567805918515 0.092150682106 -1.806977625935
O -0.100977940993 -0.878045608477 -1.546722999834
H 0.929860180854 0.791764401733 -1.017901939585
H 0.853360037419 0.338039669713 -2.857344807653
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000293692535 -0.000012596236 -0.000329785551
C 0.000140330470 -0.000037961759 -0.000239503229
O -0.000156959354 -0.000325432006 -0.000122051622
H 0.000007572095 0.000007815521 -0.000056567963
H 0.000023021425 -0.000024883470 -0.000009021896
H 0.000013975287 0.000050855610 -0.000036223695
H 0.000060999578 0.000046060921 0.000286474941
C -0.000352330542 0.000272021061 0.000249737090
O 0.000106987594 -0.000027373986 0.000156688730
H -0.000073360313 0.000005877746 0.000026694637
H -0.000039937696 0.000036397700 0.000049017340
*** Time spent in gradient calculation: 1.67 sec ***
* Info * Energy : -268.1261370352 a.u.
* Info * Gradient : 2.737549e-04 a.u. (RMS)
* Info * 5.103929e-04 a.u. (Max)
* Info * Time : 3.64 sec
Optimization Step 148
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.696756832459 1.041641387024 0.909854200983
C -0.821656147116 0.327357861705 1.633932060679
O -0.123069065102 -0.758038739408 1.250392087825
H -2.189849867547 1.905900537412 1.357045654654
H -1.963762537703 0.772838346796 -0.115759407909
H -0.590610827799 0.598989105303 2.670813230555
H -0.302457574716 -0.963046746324 0.309953975273
C 0.577709036620 0.083953672403 -1.815525754166
O -0.101975649326 -0.877299690809 -1.550561607299
H 0.949765085377 0.781920045277 -1.029435830653
H 0.864499029245 0.322876602033 -2.867257190709
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000197260253 -0.000027289432 -0.000312227730
C 0.000138723158 0.000048325030 -0.000161779932
O -0.000059555806 -0.000171792551 0.000330800549
H 0.000046102255 0.000007536021 -0.000028348865
H 0.000017707140 -0.000014825323 -0.000043847087
H 0.000017006616 0.000002364376 -0.000001803097
H -0.000041991167 -0.000095822544 -0.000201557341
C -0.000368603243 0.000195743213 0.000206140802
O 0.000134262326 -0.000055641934 0.000144425588
H -0.000049869717 0.000044442051 0.000046625636
H -0.000007567230 0.000059119565 -0.000006166912
*** Time spent in gradient calculation: 1.67 sec ***
* Info * Energy : -268.1261806632 a.u.
* Info * Gradient : 2.437545e-04 a.u. (RMS)
* Info * 4.654866e-04 a.u. (Max)
* Info * Time : 3.73 sec
Optimization Step 149
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.704594300970 1.042520280200 0.921714610364
C -0.825760749213 0.326953354188 1.639818160372
O -0.121159095650 -0.751301896280 1.247055803675
H -2.203175329744 1.899935947624 1.375935347397
H -1.968652698991 0.781431006143 -0.106695346544
H -0.596170806406 0.591222990780 2.678886814841
H -0.302623919362 -0.952603663652 0.306665249212
C 0.588405899048 0.074934802362 -1.824419643128
O -0.103541894190 -0.876128725285 -1.554566060519
H 0.972343805062 0.769935516855 -1.041384435278
H 0.875724927080 0.306605613629 -2.877523632053
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000186159297 0.000034198053 -0.000242607703
C 0.000137480768 -0.000030946716 -0.000134005914
O -0.000143819559 -0.000217524879 -0.000029554026
H 0.000019113535 0.000002134276 -0.000029088049
H 0.000026441369 -0.000008871885 -0.000021658369
H 0.000003632174 0.000024310452 -0.000020363548
H 0.000030869443 -0.000001203856 0.000143797014
C -0.000316664824 0.000236249596 0.000126438543
O 0.000148649547 -0.000105605027 0.000117487337
H -0.000057192647 0.000027505686 0.000028570508
H -0.000014834711 0.000034679010 0.000035163658
*** Time spent in gradient calculation: 1.90 sec ***
* Info * Energy : -268.1262216662 a.u.
* Info * Gradient : 2.029600e-04 a.u. (RMS)
* Info * 4.148219e-04 a.u. (Max)
* Info * Time : 4.55 sec
Optimization Step 150
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.713804213174 1.042815125613 0.934859043654
C -0.829973757372 0.326792509486 1.646040230884
O -0.118653580361 -0.743452674943 1.243261528059
H -2.217830148259 1.892892333332 1.396815676638
H -1.975327087193 0.789990735158 -0.096346455541
H -0.601299793985 0.583106000290 2.687335453662
H -0.303174315372 -0.941441321304 0.302495017077
C 0.600802948774 0.064168623767 -1.834145468790
O -0.106006613561 -0.874174873459 -1.558699910954
H 1.001003806695 0.753750891333 -1.054353973617
H 0.886876147294 0.288858262502 -2.889174128691
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000202448465 -0.000001121181 -0.000153316540
C 0.000069628332 0.000020869767 -0.000154014841
O -0.000108223752 -0.000132607680 0.000183211185
H 0.000019909127 0.000006461466 -0.000016009988
H 0.000025209711 0.000001450039 -0.000032941564
H 0.000007903827 0.000010277073 0.000010849401
H -0.000022397298 -0.000060403461 -0.000092260864
C -0.000347407877 0.000148566827 0.000108629495
O 0.000219188811 -0.000078574934 0.000105640758
H -0.000055934534 0.000034083836 0.000030105810
H 0.000004087251 0.000049537924 -0.000012075874
*** Time spent in gradient calculation: 1.71 sec ***
* Info * Energy : -268.1262625228 a.u.
* Info * Gradient : 1.905688e-04 a.u. (RMS)
* Info * 3.931472e-04 a.u. (Max)
* Info * Time : 3.88 sec
Optimization Step 151
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.723206709707 1.042377739125 0.947292972346
C -0.833706837457 0.326951417629 1.652081583389
O -0.115558779023 -0.735429381981 1.240283302546
H -2.232515029518 1.885304495449 1.416444433812
H -1.982812677845 0.796569756673 -0.086139400819
H -0.605586459153 0.576704843007 2.694981391199
H -0.303172814257 -0.930915709601 0.299854421052
C 0.613455855508 0.053072189099 -1.843185778923
O -0.109643100090 -0.871596487698 -1.563640441435
H 1.031432012936 0.734585606955 -1.065520150196
H 0.897865933857 0.272298193714 -2.899678263084
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000041662949 0.000124289345 -0.000216858151
C 0.000189031005 -0.000062027955 0.000026213888
O -0.000155047642 -0.000139690815 0.000039620715
H 0.000037489141 -0.000008975582 -0.000001268307
H 0.000026623216 0.000010750962 -0.000031855428
H -0.000014496661 -0.000007296363 -0.000040183454
H 0.000024809260 -0.000032231578 0.000085835277
C -0.000229236482 0.000236649510 -0.000023052955
O 0.000163430922 -0.000196401957 0.000062662280
H -0.000055435655 0.000047437119 0.000036169866
H -0.000017696735 0.000029053012 0.000044570056
*** Time spent in gradient calculation: 1.77 sec ***
* Info * Energy : -268.1262983724 a.u.
* Info * Gradient : 1.788594e-04 a.u. (RMS)
* Info * 3.302784e-04 a.u. (Max)
* Info * Time : 4.00 sec
Optimization Step 152
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.732004020519 1.041281413660 0.958803546801
C -0.836410626272 0.327007940538 1.656160681986
O -0.112884932461 -0.727915311433 1.234632765631
H -2.246617079309 1.877631067170 1.434065273772
H -1.991925664110 0.800440790757 -0.075765770319
H -0.606298580308 0.571498180503 2.700138113095
H -0.304363045206 -0.919692135198 0.293998905655
C 0.625470870547 0.042003921421 -1.850554570314
O -0.113522963772 -0.867763603193 -1.564576509009
H 1.062353582683 0.716198653846 -1.076989558799
H 0.907953299972 0.253508936883 -2.909381275299
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000421492112 -0.000073513637 0.000055577380
C -0.000143814215 -0.000003974064 -0.000335631539
O -0.000144546151 -0.000097605923 0.000073602212
H -0.000044454209 0.000028334985 -0.000026147354
H 0.000025362274 -0.000002581792 -0.000002232262
H 0.000027532490 0.000060268582 0.000106916737
H -0.000039214847 -0.000015069629 -0.000097026777
C -0.000507074493 0.000077672984 0.000178521275
O 0.000381537602 -0.000026563467 0.000146233117
H -0.000028639145 0.000010591981 -0.000036463940
H 0.000062726441 0.000045834579 -0.000081352330
*** Time spent in gradient calculation: 1.73 sec ***
* Info * Energy : -268.1263287271 a.u.
* Info * Gradient : 2.802686e-04 a.u. (RMS)
* Info * 5.431643e-04 a.u. (Max)
* Info * Time : 3.92 sec
Optimization Step 153
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.738229370992 1.039420393609 0.964858912515
C -0.838136086762 0.327772792343 1.659469534047
O -0.109999721153 -0.723103424400 1.234822619827
H -2.254767524515 1.872559047914 1.443494451344
H -1.997608596759 0.801196604336 -0.070459669001
H -0.608091452437 0.570239008623 2.703627670093
H -0.303828595797 -0.915805034743 0.294976201139
C 0.634235636854 0.034772664825 -1.854181833193
O -0.117989558433 -0.864818490420 -1.569962797207
H 1.085061927687 0.697219843828 -1.078319590367
H 0.914135852875 0.248578406767 -2.912987093615
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000079813207 0.000066662652 -0.000141616176
C 0.000052044178 0.000053026671 0.000025130003
O -0.000097201691 -0.000143217204 0.000170822647
H 0.000055699131 -0.000001178721 0.000003830390
H 0.000020602331 0.000022303851 -0.000022590113
H -0.000015320095 -0.000035443134 -0.000045323009
H 0.000004103580 -0.000047900707 -0.000028184542
C -0.000151155742 0.000179555973 -0.000059678024
O 0.000189782410 -0.000169842574 0.000025174970
H -0.000083016900 0.000052320021 0.000034985540
H -0.000043883350 0.000026990884 0.000022017846
*** Time spent in gradient calculation: 1.99 sec ***
* Info * Energy : -268.1263493758 a.u.
* Info * Gradient : 1.493933e-04 a.u. (RMS)
* Info * 2.559251e-04 a.u. (Max)
* Info * Time : 4.52 sec
Optimization Step 154
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.747798213541 1.036397349622 0.974573654174
C -0.839728108468 0.328384584993 1.662863002616
O -0.105925861091 -0.715582103238 1.230628412073
H -2.268297891658 1.863937448960 1.458551497505
H -2.008705760146 0.800840593378 -0.061011207246
H -0.606919571630 0.568296836860 2.706898459631
H -0.304639661143 -0.907358551128 0.291773697600
C 0.647614634199 0.023088140648 -1.858691770945
O -0.125019096496 -0.858723939961 -1.573128049438
H 1.120517504052 0.670282375780 -1.083029189018
H 0.925653519583 0.234339710727 -2.918373763739
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000137525056 0.000251868837 -0.000291475849
C 0.000261240337 -0.000138010759 0.000264049257
O -0.000129497121 -0.000140812298 0.000034561969
H 0.000071059370 -0.000007622011 0.000000457277
H 0.000013078493 0.000016487503 -0.000028537492
H -0.000028080532 -0.000044947658 -0.000096153476
H 0.000032897077 -0.000025664504 0.000094908172
C -0.000041429281 0.000309174208 -0.000145722281
O 0.000079589271 -0.000296725930 0.000056944901
H -0.000054096901 0.000079689297 0.000025427986
H -0.000054916125 -0.000000929140 0.000073013400
*** Time spent in gradient calculation: 2.04 sec ***
* Info * Energy : -268.1263806297 a.u.
* Info * Gradient : 2.385027e-04 a.u. (RMS)
* Info * 4.090345e-04 a.u. (Max)
* Info * Time : 4.54 sec
Optimization Step 155
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.753714770230 1.033510623968 0.980480688480
C -0.840591939039 0.329172065372 1.664554652731
O -0.103343487706 -0.710561183345 1.227675028706
H -2.278851626380 1.857192540992 1.466235720453
H -2.015610257623 0.797632873958 -0.054798547924
H -0.605056908310 0.568789654072 2.708438121657
H -0.305889003688 -0.901959045534 0.289405925135
C 0.656641151277 0.014993431449 -1.860287062027
O -0.130191777179 -0.853988062447 -1.575306944781
H 1.147512485492 0.647581701811 -1.083829054501
H 0.932195499080 0.228215837055 -2.920559103946
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000440831548 -0.000053725953 0.000119655637
C -0.000204468131 0.000006537416 -0.000347846912
O -0.000144385666 -0.000109027851 0.000157533582
H -0.000059180966 0.000030590781 -0.000040038770
H 0.000033230401 -0.000002558282 0.000006917889
H 0.000022109875 0.000060666593 0.000127070427
H -0.000017970178 -0.000015785078 -0.000047798746
C -0.000459392719 0.000038345364 0.000162860278
O 0.000371528755 -0.000049226840 -0.000017680324
H -0.000012301653 0.000043678814 -0.000063856156
H 0.000043057090 0.000050710684 -0.000066965069
*** Time spent in gradient calculation: 1.87 sec ***
* Info * Energy : -268.1264023359 a.u.
* Info * Gradient : 2.791480e-04 a.u. (RMS)
* Info * 4.889126e-04 a.u. (Max)
* Info * Time : 4.33 sec
Optimization Step 156
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.759834243540 1.030198435395 0.984379287044
C -0.840527263544 0.329619316360 1.664784186457
O -0.100169686050 -0.706613145693 1.224321914231
H -2.284575639490 1.852055939983 1.473474456825
H -2.024888890875 0.794654657202 -0.050145383481
H -0.602133085178 0.569354211421 2.707599961684
H -0.306093397790 -0.898305896868 0.287050190852
C 0.665241325407 0.007654282502 -1.859916952920
O -0.136369810824 -0.848415860868 -1.576012481349
H 1.168807179857 0.628104851302 -1.081810499802
H 0.940649879103 0.220094489982 -2.920045187669
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000005477124 0.000116171831 -0.000259085213
C 0.000096674968 -0.000042186419 0.000214075615
O -0.000126518124 -0.000170079799 -0.000064288586
H 0.000083652797 0.000008808963 0.000001378567
H 0.000002242926 0.000010427525 -0.000003675076
H -0.000025694667 -0.000042954427 -0.000085143676
H 0.000046917277 0.000012270810 0.000126112404
C -0.000005726575 0.000281005344 -0.000103511597
O 0.000096008194 -0.000212683788 0.000114458326
H -0.000087342101 0.000054622061 0.000003834856
H -0.000073560724 -0.000017932311 0.000048090959
*** Time spent in gradient calculation: 1.77 sec ***
* Info * Energy : -268.1264232489 a.u.
* Info * Gradient : 1.901732e-04 a.u. (RMS)
* Info * 2.995187e-04 a.u. (Max)
* Info * Time : 4.19 sec
Optimization Step 157
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.765941677372 1.026493834756 0.989514921267
C -0.840602925465 0.330399297765 1.665415738729
O -0.096989464638 -0.701869404907 1.220880020123
H -2.295037058912 1.844716109916 1.480207393331
H -2.032929035882 0.789765955434 -0.044295469246
H -0.598469676717 0.571182834000 2.707520110273
H -0.307940614938 -0.894418301848 0.284665108913
C 0.674342013867 -0.000544974411 -1.859460550716
O -0.142886217325 -0.842271823430 -1.578061820413
H 1.194792265540 0.603486895602 -1.079555713968
H 0.950014605852 0.214089408868 -2.919397665063
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000436363375 -0.000043117905 0.000110530980
C -0.000228423948 -0.000051621892 -0.000374897118
O -0.000088313836 -0.000120886423 0.000159255447
H -0.000061491885 0.000044121904 -0.000052333176
H 0.000021028922 0.000000079308 0.000003081468
H 0.000031523123 0.000064969263 0.000130202849
H -0.000029480802 -0.000016643106 -0.000066322589
C -0.000417168814 0.000073964827 0.000180059160
O 0.000302513480 -0.000044981694 0.000063070576
H -0.000005921992 0.000058198166 -0.000083098535
H 0.000050435567 0.000030694490 -0.000075862707
*** Time spent in gradient calculation: 1.83 sec ***
* Info * Energy : -268.1264464758 a.u.
* Info * Gradient : 2.719110e-04 a.u. (RMS)
* Info * 4.603498e-04 a.u. (Max)
* Info * Time : 4.22 sec
Optimization Step 158
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.771660702136 1.023608681618 0.993965868646
C -0.840730858129 0.331062162053 1.666660653714
O -0.094356445373 -0.697901971186 1.218612595914
H -2.301199000666 1.839495585148 1.487828566800
H -2.040423408687 0.787408393169 -0.039502440615
H -0.596422875451 0.571379519284 2.707944458147
H -0.308745683861 -0.891538405567 0.283555176040
C 0.682192718321 -0.007462840019 -1.859668250438
O -0.147954575658 -0.837593677920 -1.580812121282
H 1.216005740366 0.581064030481 -1.076855601359
H 0.955346582353 0.211348513767 -2.919060747718
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000061110935 0.000172968724 -0.000275555312
C 0.000192320323 -0.000086921617 0.000262653301
O -0.000134012676 -0.000118532779 0.000035870681
H 0.000067799281 -0.000008610738 -0.000005402987
H 0.000023017361 0.000003178617 -0.000024556229
H -0.000026218825 -0.000043650049 -0.000087855511
H 0.000013859049 -0.000039430762 0.000047375952
C -0.000029605560 0.000244719223 -0.000140650341
O 0.000094638512 -0.000189221891 0.000108094522
H -0.000056857419 0.000067750619 0.000018169805
H -0.000074878585 -0.000009056869 0.000057139174
*** Time spent in gradient calculation: 1.87 sec ***
* Info * Energy : -268.1264660674 a.u.
* Info * Gradient : 1.978644e-04 a.u. (RMS)
* Info * 3.369410e-04 a.u. (Max)
* Info * Time : 4.11 sec
Optimization Step 159
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.778065320556 1.019889340637 0.999834196121
C -0.841024749754 0.331700885489 1.667572960394
O -0.091096061156 -0.692900472829 1.215062890081
H -2.310952282641 1.832154636742 1.496267575245
H -2.049385231871 0.783440354532 -0.032932892246
H -0.593193766545 0.572543923292 2.708230152620
H -0.310091079856 -0.886981611807 0.281055080731
C 0.691472718650 -0.016108444832 -1.859252675657
O -0.154222315220 -0.831298445410 -1.583718513389
H 1.240712515982 0.554624170590 -1.074029253684
H 0.965644386826 0.205581017433 -2.918056452605
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000345897519 -0.000034925590 0.000015593048
C -0.000165667092 -0.000035788286 -0.000250085546
O -0.000085051131 -0.000102953279 0.000174717835
H -0.000026247415 0.000036276695 -0.000029088477
H 0.000016304692 0.000009171872 -0.000007644992
H 0.000014547103 0.000041254198 0.000097468125
H -0.000016387903 -0.000030933120 -0.000044867553
C -0.000331151356 0.000108828407 0.000107492425
O 0.000258496565 -0.000063003778 0.000044267430
H -0.000021676015 0.000037305073 -0.000055606238
H 0.000018145178 0.000027664778 -0.000056454402
*** Time spent in gradient calculation: 1.69 sec ***
* Info * Energy : -268.1264897812 a.u.
* Info * Gradient : 2.118517e-04 a.u. (RMS)
* Info * 3.647732e-04 a.u. (Max)
* Info * Time : 3.92 sec
Optimization Step 160
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.784951757824 1.016080793832 1.005467516538
C -0.840818308297 0.332435074121 1.668416220080
O -0.087901038785 -0.688025200269 1.210982624355
H -2.318950417290 1.825302521747 1.505484050845
H -2.059324547492 0.779528014620 -0.026467913719
H -0.589255107526 0.573217713013 2.707835490493
H -0.311515439367 -0.882496651206 0.278290771716
C 0.700873061902 -0.024944135316 -1.858464093038
O -0.160577640664 -0.824886901012 -1.586196070313
H 1.262583845295 0.529236854319 -1.070186150900
H 0.976604920858 0.198086674096 -2.916297677397
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000002385549 0.000125593073 -0.000246974653
C 0.000125085566 -0.000057234650 0.000219308598
O -0.000107559049 -0.000142016958 0.000022728857
H 0.000057288723 -0.000005329016 -0.000008908717
H 0.000016901339 0.000005624448 -0.000020173914
H -0.000029078631 -0.000037863242 -0.000092700455
H 0.000029836873 -0.000011711420 0.000067617400
C 0.000005706039 0.000292055486 -0.000133703238
O 0.000037252257 -0.000196370563 0.000124045910
H -0.000063528376 0.000039779783 0.000017119230
H -0.000068054878 -0.000019130803 0.000047785752
*** Time spent in gradient calculation: 1.77 sec ***
* Info * Energy : -268.1265128678 a.u.
* Info * Gradient : 1.829975e-04 a.u. (RMS)
* Info * 3.212562e-04 a.u. (Max)
* Info * Time : 3.92 sec
Optimization Step 161
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.791591090063 1.012647645882 1.011929330606
C -0.840925013689 0.333377432087 1.669369545834
O -0.085027987952 -0.682866038314 1.207089051808
H -2.328458911230 1.818200586211 1.514949942300
H -2.068755612646 0.776047705941 -0.019299719665
H -0.585286669793 0.574596860884 2.708011178309
H -0.313942566206 -0.878132723025 0.275685510096
C 0.709851005705 -0.033909653605 -1.857906634932
O -0.166162116296 -0.818947721257 -1.589523653010
H 1.284973759861 0.502355374942 -1.066888996973
H 0.987529471468 0.192365786764 -2.914800340331
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000257720597 0.000001206475 -0.000020868765
C -0.000105169956 -0.000044073188 -0.000210971239
O -0.000040308683 -0.000120666385 0.000206448530
H -0.000025701746 0.000025189619 -0.000023442400
H 0.000026057948 0.000012803258 -0.000026669839
H 0.000014064661 0.000036834396 0.000090068373
H -0.000033898013 -0.000027156599 -0.000066152228
C -0.000331473335 0.000106805624 0.000061201403
O 0.000236778781 -0.000052900323 0.000059158178
H -0.000012029443 0.000029654944 -0.000019803564
H 0.000020427685 0.000027178815 -0.000052947822
*** Time spent in gradient calculation: 1.64 sec ***
* Info * Energy : -268.1265349058 a.u.
* Info * Gradient : 1.895615e-04 a.u. (RMS)
* Info * 3.535925e-04 a.u. (Max)
* Info * Time : 3.76 sec
Optimization Step 162
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.798659272139 1.009249674786 1.018552043805
C -0.840917288085 0.334338978665 1.670512613721
O -0.082286576634 -0.677571965627 1.202731157203
H -2.336501312206 1.811548610309 1.525558257805
H -2.079411100967 0.772722888724 -0.011709983790
H -0.581234393793 0.575132758548 2.707915396322
H -0.315681637604 -0.873024554544 0.272640380409
C 0.719169609905 -0.043293034075 -1.857384931046
O -0.171612557204 -0.813084633172 -1.592624171467
H 1.304799645418 0.476814197682 -1.063312151633
H 0.999520986790 0.183969759459 -2.913087350036
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000017282680 0.000106642246 -0.000216949680
C 0.000098742049 -0.000015948007 0.000180793114
O -0.000083333611 -0.000145658067 0.000038033556
H 0.000047904922 -0.000011521919 -0.000007162802
H 0.000020503608 0.000005974825 -0.000021196223
H -0.000031476239 -0.000038356512 -0.000086645038
H 0.000034457407 -0.000009261011 0.000056559862
C -0.000009513129 0.000282199865 -0.000135898518
O 0.000023147142 -0.000186539445 0.000106214494
H -0.000050491453 0.000022911019 0.000029026680
H -0.000060433623 -0.000013884344 0.000052983785
*** Time spent in gradient calculation: 1.75 sec ***
* Info * Energy : -268.1265564211 a.u.
* Info * Gradient : 1.663078e-04 a.u. (RMS)
* Info * 3.133619e-04 a.u. (Max)
* Info * Time : 3.72 sec
Optimization Step 163
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.805424668467 1.006009759194 1.025485433508
C -0.840903289614 0.335410628574 1.671239367936
O -0.079878735725 -0.672351004215 1.198121557924
H -2.345191537997 1.805029847821 1.535800259806
H -2.090013821359 0.769381510223 -0.003741592926
H -0.576367778895 0.576816110511 2.707607018268
H -0.318725602744 -0.868038062503 0.269350986494
C 0.727951579316 -0.052665539377 -1.856838038724
O -0.176545847920 -0.807196208290 -1.595408630690
H 1.323511148977 0.452658640983 -1.060639685964
H 1.012678780898 0.174417914539 -2.911690982864
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000275872906 -0.000038435156 -0.000010972078
C -0.000148681805 -0.000021133948 -0.000253543815
O -0.000006941061 -0.000138251372 0.000146084983
H -0.000026632182 0.000032678110 -0.000019574284
H 0.000018938777 0.000017369807 -0.000020379260
H 0.000016120971 0.000040269737 0.000101635911
H -0.000025512556 -0.000002210770 -0.000029272822
C -0.000311870474 0.000104869687 0.000098547003
O 0.000203327942 -0.000040462147 0.000070539960
H -0.000016069060 0.000017772233 -0.000024645601
H 0.000029003766 0.000026114542 -0.000063844423
*** Time spent in gradient calculation: 1.70 sec ***
* Info * Energy : -268.1265762950 a.u.
* Info * Gradient : 1.894278e-04 a.u. (RMS)
* Info * 3.434710e-04 a.u. (Max)
* Info * Time : 3.83 sec
Optimization Step 164
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.812001976810 1.003363451715 1.032286927485
C -0.841010379764 0.336548272640 1.672834486617
O -0.078005412307 -0.667384808711 1.194442270263
H -2.352747666346 1.799206082374 1.546293284355
H -2.099655469592 0.766744664515 0.003890257716
H -0.572547262412 0.577332714781 2.707955876507
H -0.320994450762 -0.863404130911 0.266862041608
C 0.736266840434 -0.061378756136 -1.857234357813
O -0.180419191547 -0.802448970483 -1.598888194151
H 1.340406056259 0.429671367136 -1.058478176409
H 1.023147913067 0.166820543630 -2.910963400874
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000080375162 0.000173886216 -0.000220570185
C 0.000189491706 -0.000051526916 0.000224422221
O -0.000054621194 -0.000123659411 0.000090295806
H 0.000036427633 -0.000025251158 -0.000014965506
H 0.000029942244 -0.000000198483 -0.000033165957
H -0.000029923780 -0.000042013006 -0.000103555701
H 0.000008507009 -0.000029308014 -0.000004727255
C 0.000008541309 0.000257805630 -0.000145369975
O -0.000023439423 -0.000191056450 0.000093688689
H -0.000018997988 0.000035871357 0.000044395201
H -0.000057586340 -0.000004500701 0.000063184017
*** Time spent in gradient calculation: 1.80 sec ***
* Info * Energy : -268.1265932706 a.u.
* Info * Gradient : 1.818273e-04 a.u. (RMS)
* Info * 2.982071e-04 a.u. (Max)
* Info * Time : 4.00 sec
Optimization Step 165
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.817765985775 1.000714644044 1.038664680551
C -0.841303166836 0.337462255789 1.673644334042
O -0.076289189020 -0.662757174146 1.190485096220
H -2.359389121569 1.794042009422 1.555850796221
H -2.109294749134 0.764345248270 0.011311544479
H -0.568806750990 0.578536152170 2.707988656217
H -0.323219833006 -0.858306618387 0.263895330625
C 0.743600273045 -0.069578769959 -1.857288863531
O -0.183860760510 -0.797656642052 -1.601317145223
H 1.354240227180 0.410780237615 -1.057127444543
H 1.035124737778 0.156661351004 -2.910446469437
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000302222799 -0.000103599592 -0.000022395852
C -0.000196448503 0.000039161932 -0.000240655872
O -0.000023670557 -0.000127865311 0.000056884678
H -0.000004823097 0.000036008140 -0.000006205121
H 0.000008145977 0.000017243598 -0.000003608701
H 0.000011461389 0.000030486382 0.000095163129
H 0.000007814771 0.000013493059 0.000031758510
C -0.000266557336 0.000075949246 0.000123203791
O 0.000182749894 -0.000019096359 0.000047518207
H -0.000031102264 0.000009542156 -0.000033965863
H 0.000018720890 0.000030209962 -0.000055371299
*** Time spent in gradient calculation: 1.60 sec ***
* Info * Energy : -268.1266079352 a.u.
* Info * Gradient : 1.830114e-04 a.u. (RMS)
* Info * 3.202703e-04 a.u. (Max)
* Info * Time : 3.65 sec
Optimization Step 166
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.823929726827 0.998364600964 1.045294536101
C -0.841356663123 0.338518450242 1.675126537026
O -0.074826078235 -0.658179557182 1.186908270774
H -2.366805585991 1.788573940278 1.565728674999
H -2.118381663034 0.761752747516 0.018774299697
H -0.564778381498 0.579269399167 2.708136842184
H -0.325955021110 -0.853857654836 0.261384394970
C 0.751244332748 -0.077826616560 -1.858050525386
O -0.186988556205 -0.793090262803 -1.604266002423
H 1.368574761538 0.391107234451 -1.056113063750
H 1.045791946930 0.147178378458 -2.910390656574
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000075607462 0.000185241789 -0.000177342260
C 0.000184628502 -0.000096281034 0.000165729711
O -0.000014211710 -0.000107344223 0.000118473522
H 0.000012945879 -0.000018487496 -0.000027970818
H 0.000023330976 -0.000003885499 -0.000030690602
H -0.000019719248 -0.000027169449 -0.000089688172
H -0.000017664217 -0.000028909482 -0.000038977093
C 0.000028531914 0.000244299308 -0.000092759279
O -0.000066553307 -0.000182363927 0.000088214786
H -0.000003307954 0.000040479900 0.000030472509
H -0.000043170006 -0.000003334446 0.000045079230
*** Time spent in gradient calculation: 1.65 sec ***
* Info * Energy : -268.1266214919 a.u.
* Info * Gradient : 1.661737e-04 a.u. (RMS)
* Info * 2.673598e-04 a.u. (Max)
* Info * Time : 3.75 sec
Optimization Step 167
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.829722157001 0.995925185575 1.052166767965
C -0.842007845542 0.339466953080 1.676563855619
O -0.073489385101 -0.653382622929 1.183424454529
H -2.373252034660 1.783447919459 1.576157239619
H -2.127494019130 0.760191349941 0.026424537342
H -0.561947359862 0.579824610134 2.709032147562
H -0.327934806890 -0.848460602166 0.258788340823
C 0.758388047623 -0.086038838801 -1.859102869684
O -0.189503818742 -0.788754787227 -1.607236593957
H 1.382123879308 0.372017699371 -1.055943750429
H 1.056452378202 0.137872566174 -2.910930450393
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000256606512 -0.000111653321 -0.000040683523
C -0.000172817614 0.000061661852 -0.000199112459
O -0.000033015149 -0.000105773183 0.000035162087
H 0.000007401326 0.000029091231 0.000004226766
H 0.000012074120 0.000013533749 -0.000003952313
H 0.000014165373 0.000027228198 0.000094647549
H 0.000008153945 0.000002645767 0.000030776362
C -0.000285502413 0.000015236021 0.000117838682
O 0.000206227544 0.000020500556 0.000025138076
H -0.000023357635 0.000010371690 -0.000024963368
H 0.000019947210 0.000040449725 -0.000049866730
*** Time spent in gradient calculation: 1.78 sec ***
* Info * Energy : -268.1266337559 a.u.
* Info * Gradient : 1.716206e-04 a.u. (RMS)
* Info * 3.092406e-04 a.u. (Max)
* Info * Time : 3.87 sec
Optimization Step 168
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.835344414558 0.993809568259 1.058443739000
C -0.842225440847 0.340250758676 1.677934218849
O -0.072202908884 -0.649092932056 1.180006044613
H -2.380037364456 1.778360212631 1.585510408433
H -2.136107805308 0.757941581276 0.033564403589
H -0.558577549635 0.580101785293 2.709216993156
H -0.329984012411 -0.843637220058 0.256108050021
C 0.765292665507 -0.093679683208 -1.860036532818
O -0.192024664233 -0.784513093229 -1.609746362639
H 1.393542417912 0.355987700991 -1.055607078777
H 1.067194256743 0.126627009160 -2.911277346577
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000037080376 0.000150790772 -0.000151641448
C 0.000143121278 -0.000082959304 0.000137315147
O -0.000014169729 -0.000092521317 0.000089173262
H 0.000012695478 -0.000011814074 -0.000029995249
H 0.000007675155 -0.000007720789 -0.000012620720
H -0.000021211520 -0.000028063759 -0.000094036622
H 0.000000541566 -0.000015389277 -0.000020282930
C 0.000088233739 0.000250074654 -0.000068421181
O -0.000112547028 -0.000176776753 0.000073348925
H -0.000011365340 0.000029468219 0.000016380511
H -0.000045549697 -0.000011326786 0.000047983605
*** Time spent in gradient calculation: 1.87 sec ***
* Info * Energy : -268.1266443691 a.u.
* Info * Gradient : 1.514541e-04 a.u. (RMS)
* Info * 2.738686e-04 a.u. (Max)
* Info * Time : 3.74 sec
Optimization Step 169
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.840764746980 0.991699187198 1.065157106530
C -0.842941746955 0.341120249085 1.679576483286
O -0.071238068971 -0.644603003019 1.176896075600
H -2.386305970006 1.773448751370 1.595636725272
H -2.143935983100 0.757120496205 0.040680202665
H -0.556168693893 0.580329434167 2.710436185192
H -0.332398470529 -0.838704443829 0.253903280992
C 0.771807632455 -0.101371886432 -1.861677267326
O -0.193821976635 -0.780547718336 -1.612560564627
H 1.405969544776 0.338507790176 -1.056483337803
H 1.076520097055 0.117725800723 -2.912615135172
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000165126449 -0.000061453021 -0.000046964603
C -0.000096617080 0.000024295474 -0.000154288138
O -0.000010649597 -0.000092496295 0.000052790004
H 0.000000965701 0.000021172688 0.000000328209
H 0.000017989136 0.000011148835 -0.000014563393
H 0.000018122597 0.000027732058 0.000080948712
H -0.000018541214 -0.000010509914 0.000005927906
C -0.000262807803 0.000007779991 0.000092462790
O 0.000181428249 0.000023663967 0.000028050657
H -0.000008077467 0.000014399923 -0.000011485341
H 0.000024116347 0.000038647163 -0.000047655557
*** Time spent in gradient calculation: 1.78 sec ***
* Info * Energy : -268.1266539824 a.u.
* Info * Gradient : 1.363069e-04 a.u. (RMS)
* Info * 2.787074e-04 a.u. (Max)
* Info * Time : 3.92 sec
Optimization Step 170
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.846391287911 0.989594113779 1.071841473832
C -0.843544295018 0.341737912886 1.681233272302
O -0.070025731840 -0.639994749693 1.173477663080
H -2.392956672195 1.768168443159 1.605801281170
H -2.152595226337 0.755432858522 0.048168157120
H -0.553717833058 0.579784941016 2.711233246701
H -0.333589667269 -0.832882308267 0.250909126610
C 0.778633350491 -0.109189876619 -1.863038391501
O -0.195874008153 -0.776437193840 -1.615272759548
H 1.416798609954 0.322810278719 -1.056766964955
H 1.087185378673 0.105796052927 -2.913464855135
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000017736905 0.000079939605 -0.000125874513
C 0.000074464834 -0.000028923244 0.000099475351
O -0.000035100586 -0.000074757240 0.000043744211
H 0.000021424520 -0.000006038795 -0.000020815857
H 0.000000138665 -0.000009598341 0.000002389943
H -0.000020089140 -0.000026191267 -0.000074622752
H 0.000027482199 -0.000007728361 -0.000000236977
C 0.000063430747 0.000198182678 -0.000043212478
O -0.000079872660 -0.000126594180 0.000049402834
H -0.000017867973 0.000015861407 0.000007311886
H -0.000040329498 -0.000009429906 0.000046068874
*** Time spent in gradient calculation: 1.78 sec ***
* Info * Energy : -268.1266630688 a.u.
* Info * Gradient : 1.089353e-04 a.u. (RMS)
* Info * 2.125256e-04 a.u. (Max)
* Info * Time : 4.01 sec
Optimization Step 171
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.852080432229 0.987639545096 1.079140463344
C -0.844385107026 0.342532932137 1.683173428570
O -0.069175760459 -0.635264918359 1.170280516592
H -2.399938666635 1.762871142885 1.616727267246
H -2.160363751292 0.755210229904 0.055645169034
H -0.551401781565 0.579611129222 2.712724213688
H -0.336591886244 -0.827637260440 0.248689565872
C 0.785396640562 -0.117305457560 -1.865263035495
O -0.197391627453 -0.772305328773 -1.618247063662
H 1.429003002760 0.305674384097 -1.058491467396
H 1.097075296494 0.095011743269 -2.915530882115
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000079431034 0.000001619549 -0.000039157040
C -0.000017854591 -0.000029950974 -0.000113520959
O 0.000022092424 -0.000078445169 0.000086255071
H -0.000012245404 0.000014859745 -0.000010357225
H 0.000016697258 0.000010394522 -0.000022759193
H 0.000016742857 0.000023824927 0.000051969813
H -0.000043458520 -0.000016215581 -0.000018103172
C -0.000171033210 0.000047251410 0.000057468966
O 0.000099560741 -0.000010344378 0.000032169094
H 0.000001915224 0.000013935500 -0.000002086295
H 0.000020218383 0.000028013976 -0.000040344024
*** Time spent in gradient calculation: 1.90 sec ***
* Info * Energy : -268.1266712552 a.u.
* Info * Gradient : 9.160490e-05 a.u. (RMS)
* Info * 1.865147e-04 a.u. (Max)
* Info * Time : 4.10 sec
Optimization Step 172
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.857957244766 0.985267864384 1.086403217893
C -0.845225391245 0.343113424184 1.684978598348
O -0.068037830684 -0.630163975171 1.166418915948
H -2.406433905740 1.757177347900 1.628053080177
H -2.169474445187 0.753768934710 0.063717666237
H -0.549269257837 0.578570698167 2.713892788897
H -0.337375479875 -0.820704547227 0.245115352194
C 0.792401963593 -0.125764197466 -1.866808551427
O -0.199180990245 -0.767927972156 -1.620943520579
H 1.439846173005 0.289428142126 -1.059133030642
H 1.108197588015 0.081768395942 -2.916638342216
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000092628319 -0.000033209932 -0.000101687298
C -0.000032316859 0.000051767663 0.000038448809
O -0.000065560225 -0.000067731775 -0.000054993311
H 0.000037877288 0.000003430069 -0.000001259879
H -0.000000353936 -0.000007248162 0.000014656402
H -0.000012712470 -0.000016262578 -0.000028566836
H 0.000059011441 0.000008021914 0.000050701039
C -0.000035069448 0.000085806259 0.000006877435
O 0.000021268353 -0.000027264192 0.000031645649
H -0.000027652523 0.000007418207 -0.000004608509
H -0.000024595714 0.000000337531 0.000028412398
*** Time spent in gradient calculation: 1.90 sec ***
* Info * Energy : -268.1266789553 a.u.
* Info * Gradient : 7.342343e-05 a.u. (RMS)
* Info * 1.415034e-04 a.u. (Max)
* Info * Time : 3.90 sec
Optimization Step 173
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.863299452797 0.984126844984 1.093877257840
C -0.846385131094 0.343647344032 1.687578109061
O -0.067441497637 -0.625567975268 1.164031692789
H -2.414095861589 1.751938120319 1.638976473772
H -2.175949266113 0.754777113311 0.070941910663
H -0.548066626696 0.577273662575 2.716222109712
H -0.340261640148 -0.815589313850 0.243408237519
C 0.798810801606 -0.133333219215 -1.870048622721
O -0.199954566271 -0.764590283827 -1.624568511754
H 1.451261748868 0.273641383458 -1.062097580978
H 1.116777616932 0.071581128857 -2.919783954820
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000081799166 0.000174057700 -0.000041935024
C 0.000163179928 -0.000136739770 0.000031075300
O 0.000064109710 -0.000036651510 0.000219872903
H -0.000035901464 -0.000009630520 -0.000038948655
H 0.000011043057 -0.000002381860 -0.000033018536
H -0.000000754850 -0.000001886383 -0.000041749788
H -0.000073351890 -0.000043873555 -0.000122194916
C 0.000056992686 0.000182765017 -0.000048528240
O -0.000110039068 -0.000137342692 0.000024569736
H 0.000028407131 0.000013775052 0.000019569101
H -0.000009057198 0.000002415740 0.000008953995
*** Time spent in gradient calculation: 1.86 sec ***
* Info * Energy : -268.1266846609 a.u.
* Info * Gradient : 1.474215e-04 a.u. (RMS)
* Info * 2.319428e-04 a.u. (Max)
* Info * Time : 3.93 sec
Optimization Step 174
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.866305209097 0.982625405287 1.097653333527
C -0.846786284062 0.344052069123 1.688192652099
O -0.066989489478 -0.622845272870 1.161521929315
H -2.416320316412 1.749289049358 1.645236339679
H -2.181085253981 0.754249367727 0.075230041498
H -0.546685558237 0.577053529258 2.716608139799
H -0.340867052518 -0.811515013874 0.241235848710
C 0.802348293594 -0.138054545294 -1.870605338439
O -0.200856026030 -0.761966962260 -1.625410030225
H 1.456144640894 0.266047829118 -1.062424296662
H 1.123242463558 0.063063667388 -2.920292890877
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000149752423 -0.000125536837 -0.000047437289
C -0.000128906968 0.000077999551 -0.000104538158
O -0.000036989091 -0.000079791505 -0.000092269708
H 0.000026976160 0.000021222571 0.000014986480
H 0.000007416382 0.000011797866 0.000003498171
H 0.000010117369 0.000015599481 0.000056623029
H 0.000028886623 0.000021805845 0.000097744449
C -0.000171500814 -0.000030016628 0.000079623532
O 0.000145965222 0.000063616080 0.000018335774
H -0.000030667302 0.000003968722 -0.000016519217
H 0.000012236010 0.000023943842 -0.000033123704
*** Time spent in gradient calculation: 1.91 sec ***
* Info * Energy : -268.1266880392 a.u.
* Info * Gradient : 1.249473e-04 a.u. (RMS)
* Info * 2.010860e-04 a.u. (Max)
* Info * Time : 3.92 sec
Optimization Step 175
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.869488866957 0.981755470674 1.101840436567
C -0.847244682780 0.344321282993 1.689413050481
O -0.066513995571 -0.620136462741 1.159729511898
H -2.420990837188 1.745933312128 1.651312500986
H -2.185456102797 0.753783767859 0.079628461376
H -0.545467342478 0.576390933924 2.717398902864
H -0.342093556567 -0.808327096253 0.239612341642
C 0.806118774527 -0.142561336013 -1.871882396955
O -0.201561108512 -0.759757705072 -1.627213942842
H 1.462602824726 0.256596805886 -1.063338416225
H 1.128524652066 0.056550332651 -2.921364527842
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000033109479 0.000111564270 -0.000048284434
C 0.000104490314 -0.000088807844 0.000031085503
O 0.000024255776 -0.000031260168 0.000121786043
H -0.000015986903 -0.000006696916 -0.000028598066
H 0.000004558902 -0.000008313514 -0.000014933537
H -0.000003419092 -0.000004979859 -0.000037848699
H -0.000031327476 -0.000026015312 -0.000081690928
C 0.000041020014 0.000134240705 -0.000025733540
O -0.000079804424 -0.000085667702 0.000034175229
H 0.000015824150 0.000013693721 0.000009059112
H -0.000013009150 -0.000003856418 0.000016508311
*** Time spent in gradient calculation: 1.91 sec ***
* Info * Energy : -268.1266908933 a.u.
* Info * Gradient : 9.510400e-05 a.u. (RMS)
* Info * 1.427075e-04 a.u. (Max)
* Info * Time : 4.25 sec
Optimization Step 176
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.872944516012 0.980638385789 1.106727118797
C -0.848150343225 0.344716668090 1.691027512279
O -0.066157725095 -0.616825180510 1.157777469075
H -2.424481496478 1.742738592904 1.659076801926
H -2.190272643490 0.754481903577 0.084575519239
H -0.545080980131 0.575204262396 2.719042803411
H -0.342808245945 -0.803745066242 0.237931124375
C 0.810200306082 -0.147822146616 -1.873679984694
O -0.201993089135 -0.757507644827 -1.629380345566
H 1.468689074485 0.247226340638 -1.064809401317
H 1.134924815905 0.048448915886 -2.923036090303
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000084450779 -0.000069046418 -0.000062029353
C -0.000061069353 0.000067470916 -0.000006194446
O -0.000046145299 -0.000054311281 -0.000058616317
H 0.000031145863 0.000006277610 0.000009394086
H 0.000005214568 0.000003044650 0.000004782734
H -0.000002806123 -0.000004885677 0.000005629636
H 0.000042271037 0.000009051053 0.000061985382
C -0.000080946656 0.000010941163 0.000023899956
O 0.000069114616 0.000018883963 0.000001391029
H -0.000022672607 0.000002067843 -0.000005785772
H -0.000004627112 0.000013947016 0.000000476245
*** Time spent in gradient calculation: 1.96 sec ***
* Info * Energy : -268.1266938827 a.u.
* Info * Gradient : 6.919023e-05 a.u. (RMS)
* Info * 1.254870e-04 a.u. (Max)
* Info * Time : 3.98 sec
Optimization Step 177
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.876875120344 0.979329807106 1.111892389430
C -0.848517926213 0.345184632465 1.692069771789
O -0.065659589754 -0.613533918063 1.155065814987
H -2.429512798616 1.738849345224 1.666717110226
H -2.196174261516 0.753958360462 0.090114992755
H -0.542820728450 0.575163914849 2.719474164147
H -0.345249265483 -0.799897015656 0.235846332510
C 0.814856645399 -0.153736201978 -1.874901730930
O -0.202932087294 -0.754321736383 -1.631066302747
H 1.476212917708 0.236006293513 -1.065757794715
H 1.142234298202 0.038791090798 -2.924182810685
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000018390555 0.000037155873 -0.000004591310
C 0.000025675184 -0.000074301940 -0.000088888629
O 0.000048902218 -0.000041772169 0.000077278478
H -0.000025364027 0.000008823333 -0.000017322315
H 0.000006295470 0.000006324116 -0.000019515067
H 0.000013073271 0.000019048152 0.000025238510
H -0.000052843333 -0.000008889822 -0.000033979541
C -0.000035817467 0.000057077651 0.000032173148
O -0.000002500207 -0.000019061383 0.000034437973
H 0.000007627976 0.000010168859 -0.000002673751
H 0.000010960854 0.000007948874 -0.000028930440
*** Time spent in gradient calculation: 2.02 sec ***
* Info * Energy : -268.1266966549 a.u.
* Info * Gradient : 6.099798e-05 a.u. (RMS)
* Info * 1.186642e-04 a.u. (Max)
* Info * Time : 4.52 sec
Optimization Step 178
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.880327940430 0.978682596338 1.117228080594
C -0.849748497877 0.345347020349 1.694333262058
O -0.065487931222 -0.610020417145 1.153371118254
H -2.433655972780 1.735435048568 1.675025901639
H -2.200607089895 0.755079229865 0.095401011202
H -0.543282659633 0.572656436711 2.721900137805
H -0.345042405582 -0.794524295766 0.233805901652
C 0.819004457950 -0.158914924420 -1.877474062153
O -0.202895028285 -0.752605047825 -1.633681542408
H 1.482583023861 0.226786025342 -1.068205805235
H 1.147837249589 0.030890110045 -2.926610527270
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000024088843 0.000060721683 -0.000078876003
C 0.000067741761 0.000006815322 0.000148620586
O -0.000047492607 -0.000013011603 0.000020193357
H 0.000025399132 -0.000017278692 -0.000011921698
H 0.000000477953 -0.000018374984 0.000007693894
H -0.000022411286 -0.000031755774 -0.000079123668
H 0.000045788765 -0.000014219988 -0.000031986540
C 0.000074741139 0.000106161511 -0.000065399183
O -0.000073122851 -0.000069467323 -0.000003900432
H 0.000001747067 0.000002490800 0.000009676476
H -0.000034122862 -0.000010543796 0.000057754772
*** Time spent in gradient calculation: 1.92 sec ***
* Info * Energy : -268.1266989802 a.u.
* Info * Gradient : 8.977571e-05 a.u. (RMS)
* Info * 1.634732e-04 a.u. (Max)
* Info * Time : 3.92 sec
Optimization Step 179
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.882635730096 0.977663134235 1.120169808474
C -0.849883392172 0.345789899542 1.694671744692
O -0.065193987413 -0.608099925558 1.151673032097
H -2.435923741049 1.733361452897 1.679566602126
H -2.204270712968 0.754958848818 0.098553794444
H -0.541666091324 0.573279396558 2.721912353122
H -0.347011183460 -0.792493102367 0.232867216931
C 0.821618374231 -0.162556561082 -1.877895837926
O -0.203523258302 -0.750547786810 -1.634553385969
H 1.486346440702 0.220500624534 -1.068354174312
H 1.152718951051 0.024670923450 -2.927013894735
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000084266904 -0.000074964856 -0.000007620640
C -0.000077658125 0.000022426660 -0.000110252396
O 0.000005060209 -0.000050561022 -0.000032249094
H 0.000003082141 0.000016657783 0.000007348518
H 0.000006542083 0.000014092624 -0.000008221072
H 0.000015238852 0.000018630934 0.000054234438
H -0.000005737903 0.000012654126 0.000052189996
C -0.000114824623 -0.000029336018 0.000062004764
O 0.000093799819 0.000048166183 0.000008609748
H -0.000014785312 0.000003417446 -0.000011825356
H 0.000020196552 0.000019952132 -0.000042282522
*** Time spent in gradient calculation: 1.97 sec ***
* Info * Energy : -268.1267002781 a.u.
* Info * Gradient : 8.214395e-05 a.u. (RMS)
* Info * 1.367089e-04 a.u. (Max)
* Info * Time : 4.19 sec
Optimization Step 180
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.884858584244 0.977147405316 1.123349109700
C -0.850317545259 0.346000042394 1.695697732359
O -0.065027782894 -0.605997550560 1.150291732819
H -2.438829555580 1.731102997203 1.684322145975
H -2.207522061192 0.754773771172 0.101974487624
H -0.541026200204 0.572417457221 2.722682219219
H -0.347402112435 -0.789567449225 0.231384166324
C 0.824310273884 -0.166009169552 -1.878943501966
O -0.203830674288 -0.749095837131 -1.635899479694
H 1.490655056723 0.213835898078 -1.069177907010
H 1.156472229857 0.019380006323 -2.927889844220
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000020694848 0.000054507815 -0.000041603422
C 0.000059439003 -0.000026635924 0.000063805938
O -0.000010237672 -0.000013722658 0.000037781120
H 0.000004992460 -0.000009751718 -0.000014060497
H 0.000002101828 -0.000010084848 -0.000002031842
H -0.000009721462 -0.000013298456 -0.000043319821
H 0.000007661325 -0.000014040414 -0.000038216659
C 0.000046803906 0.000077052157 -0.000033454738
O -0.000054476849 -0.000042562805 0.000009566704
H 0.000006831151 0.000005556735 0.000005116600
H -0.000017371823 -0.000006643618 0.000027664609
*** Time spent in gradient calculation: 1.82 sec ***
* Info * Energy : -268.1267014793 a.u.
* Info * Gradient : 5.623762e-05 a.u. (RMS)
* Info * 9.616059e-05 a.u. (Max)
* Info * Time : 4.07 sec
Optimization Step 181
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.887465396871 0.976380573679 1.127097678258
C -0.850867319860 0.346351159804 1.696732759485
O -0.064845583054 -0.603560250216 1.148704154551
H -2.441657013070 1.728706008000 1.690106513295
H -2.211252247437 0.755361663940 0.105777009023
H -0.540234412400 0.571949050330 2.723612199891
H -0.348771208126 -0.786454413921 0.230211701197
C 0.827315845187 -0.170066284035 -1.880197912590
O -0.204086139422 -0.747315268429 -1.637420804044
H 1.495042133322 0.206889539863 -1.070246149422
H 1.161388737282 0.012732300736 -2.929114371218
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000043675972 -0.000031147921 -0.000009490094
C -0.000031701121 -0.000000209805 -0.000059521229
O 0.000006175882 -0.000031452888 -0.000003371982
H 0.000000096175 0.000008855097 0.000000405268
H 0.000003982634 0.000006878377 -0.000006864200
H 0.000008597184 0.000009182350 0.000026364978
H -0.000006091364 0.000004688380 0.000019178829
C -0.000056344715 0.000000594205 0.000030356885
O 0.000044041642 0.000019869303 0.000003635942
H -0.000007056811 0.000001616779 -0.000006533490
H 0.000010445903 0.000010881568 -0.000023531699
*** Time spent in gradient calculation: 2.01 sec ***
* Info * Energy : -268.1267026613 a.u.
* Info * Gradient : 3.970827e-05 a.u. (RMS)
* Info * 6.743724e-05 a.u. (Max)
* Info * Time : 4.17 sec
Optimization Step 182
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.890426612327 0.975538036960 1.131325254721
C -0.851349612423 0.346772423340 1.697893123376
O -0.064646965696 -0.600749781282 1.146706474349
H -2.445010308327 1.725861606059 1.696559070562
H -2.215807764034 0.755202231791 0.110363852434
H -0.539038476629 0.571286798930 2.724405425272
H -0.349695271356 -0.782653878850 0.228343197525
C 0.830880385584 -0.174896227321 -1.881358694073
O -0.204541827740 -0.745213742417 -1.639096849871
H 1.500474530145 0.197960443550 -1.071037075822
H 1.166822986624 0.005175846035 -2.930059850722
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000001540470 0.000010717943 -0.000031388730
C 0.000017605394 0.000004006273 0.000041324530
O -0.000018621184 -0.000014900389 -0.000007474387
H 0.000011332792 -0.000006086153 -0.000004946109
H 0.000002613546 -0.000006179753 0.000000748707
H -0.000006056260 -0.000009351481 -0.000026244476
H 0.000017186206 -0.000005788607 -0.000005945001
C 0.000015216384 0.000035385863 -0.000017968417
O -0.000014767931 -0.000009281979 0.000003597056
H 0.000000690271 0.000002563204 0.000001017613
H -0.000010516945 -0.000002222618 0.000017125362
*** Time spent in gradient calculation: 1.84 sec ***
* Info * Energy : -268.1267038601 a.u.
* Info * Gradient : 2.648180e-05 a.u. (RMS)
* Info * 4.509675e-05 a.u. (Max)
* Info * Time : 4.00 sec
Optimization Step 183
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.893256229437 0.975334888939 1.135888271282
C -0.852332829830 0.346964556242 1.699587888725
O -0.064668689582 -0.597927248935 1.145318700485
H -2.448820206594 1.723257781986 1.703385566310
H -2.219202104085 0.757140247750 0.114597362629
H -0.539057529161 0.569797379274 2.726251432273
H -0.351326589085 -0.778851594450 0.227189643774
C 0.834167036554 -0.179164797291 -1.883814823115
O -0.204180669085 -0.743932710779 -1.641024264371
H 1.505113002199 0.192144372608 -1.073885135928
H 1.171753698887 -0.003029834141 -2.932745781380
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000018731195 0.000051647029 0.000024081634
C 0.000040800555 -0.000087153358 -0.000057332910
O 0.000058620867 -0.000009450057 0.000100174901
H -0.000029226315 0.000006375674 -0.000018667260
H 0.000001324988 0.000006383128 -0.000018458790
H 0.000009515358 0.000012169628 0.000012587795
H -0.000056277617 -0.000007407315 -0.000050799171
C 0.000028507768 0.000056810514 0.000001260456
O -0.000038212805 -0.000033321994 0.000002501399
H 0.000010719045 0.000002112712 -0.000002035652
H 0.000009775728 -0.000000173515 -0.000024173232
*** Time spent in gradient calculation: 1.97 sec ***
* Info * Energy : -268.1267046095 a.u.
* Info * Gradient : 6.392793e-05 a.u. (RMS)
* Info * 1.164505e-04 a.u. (Max)
* Info * Time : 4.30 sec
Optimization Step 184
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.962151378634 0.969653054081 1.195335773791
C -0.845797055696 0.438077791977 1.707273353729
O -0.031410118649 -0.470880717534 1.130117412844
H -2.529893896430 1.698722141566 1.776143306625
H -2.333299076845 0.699859913566 0.202357584288
H -0.484841282546 0.715929149833 2.706402193688
H -0.359954900436 -0.688160275647 0.235152923202
C 0.848846181142 -0.230352215245 -1.849209252680
O -0.255419173343 -0.662848868337 -1.635966931084
H 1.538854619906 0.047201103124 -1.018652104557
H 1.232977125352 -0.084387138502 -2.888690335896
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.001384933107 -0.001596815338 0.000219168737
C -0.001604064213 0.000712929444 -0.002014601343
O -0.000156755403 -0.000012100869 -0.000685131619
H -0.000002116341 0.000244809102 0.000280615548
H 0.000007683012 0.000158699514 0.000003899520
H 0.000355205220 0.000341790728 0.001160991044
H 0.000479958736 0.000004552208 0.000557085708
C -0.003132140109 -0.001733258191 0.001576202731
O 0.002354276322 0.001640227254 -0.000321721412
H -0.000312265580 -0.000056540538 -0.000072924440
H 0.000652636196 0.000285826854 -0.000737495953
*** Time spent in gradient calculation: 1.80 sec ***
* Info * Energy : -268.1265801238 a.u.
* Info * Gradient : 1.888684e-03 a.u. (RMS)
* Info * 3.911381e-03 a.u. (Max)
* Info * Time : 4.50 sec
Optimization Step 185
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.927232333602 0.966360287794 1.161025235432
C -0.849316940220 0.378152234314 1.695463733786
O -0.049763153666 -0.545971705097 1.122500403180
H -2.487467125927 1.701217925926 1.741339332343
H -2.276159866619 0.732832547179 0.150859955211
H -0.511730592274 0.618088144580 2.711472612008
H -0.357570706601 -0.736309036598 0.213651181909
C 0.844932638365 -0.197739682775 -1.875255173958
O -0.228025622931 -0.701135641532 -1.652865139841
H 1.524915208870 0.122894653794 -1.051590512802
H 1.208092632631 -0.028227327567 -2.917699417995
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000618146378 -0.000802255079 0.000120922698
C -0.000768766634 0.000402095439 -0.001059525590
O -0.000038720487 -0.000110668225 -0.000375854055
H -0.000003077733 0.000119744779 0.000145918158
H 0.000017397326 0.000085386710 -0.000006141345
H 0.000179587694 0.000148845765 0.000598492549
H 0.000220347707 0.000133695941 0.000288241824
C -0.001525653894 -0.000927566735 0.000858020801
O 0.001059163414 0.000769782030 -0.000157794454
H -0.000070316564 -0.000008117437 -0.000066268259
H 0.000334939007 0.000184602182 -0.000381804416
*** Time spent in gradient calculation: 1.80 sec ***
* Info * Energy : -268.1266810360 a.u.
* Info * Gradient : 9.361887e-04 a.u. (RMS)
* Info * 1.980959e-03 a.u. (Max)
* Info * Time : 4.35 sec
Optimization Step 186
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.910365451842 0.969697328968 1.147700597143
C -0.851397385700 0.359080521089 1.695894385305
O -0.057933421699 -0.574452206066 1.130239290411
H -2.467997423468 1.709980774775 1.723353361007
H -2.247505005827 0.746932024942 0.131091129266
H -0.526303016618 0.587308848346 2.718148580280
H -0.354745331807 -0.757561109907 0.216049747305
C 0.840502765347 -0.186398349605 -1.881883648704
O -0.215315746198 -0.721914535360 -1.650426548855
H 1.515742302613 0.159402638474 -1.064380019733
H 1.191260141020 -0.011941965561 -2.927234966058
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000288725767 -0.000395041182 0.000062567344
C -0.000372198945 0.000202083274 -0.000533314615
O -0.000008960603 -0.000087629711 -0.000190708489
H 0.000000130125 0.000060082905 0.000070439108
H 0.000013448365 0.000044537369 -0.000007005466
H 0.000088461114 0.000068266626 0.000297078461
H 0.000102814506 0.000100357548 0.000144362848
C -0.000734263764 -0.000460721650 0.000438337708
O 0.000494342075 0.000359924670 -0.000081100293
H -0.000016743543 0.000004387977 -0.000041327328
H 0.000164180182 0.000100443109 -0.000193581593
*** Time spent in gradient calculation: 1.82 sec ***
* Info * Energy : -268.1267016411 a.u.
* Info * Gradient : 4.577452e-04 a.u. (RMS)
* Info * 9.713638e-04 a.u. (Max)
* Info * Time : 4.14 sec
Optimization Step 187
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.901942288485 0.972366668033 1.141929939255
C -0.852153627103 0.352230834598 1.697597536401
O -0.061524160090 -0.586552010529 1.136933255239
H -2.458410024013 1.716104436029 1.714118844350
H -2.233342254967 0.752882950704 0.122711603247
H -0.533187218129 0.576679763871 2.722361882401
H -0.352935271788 -0.767784333872 0.220538896305
C 0.837622783085 -0.182395584481 -1.883584203176
O -0.209451372664 -0.732838627284 -1.646484901169
H 1.510601482941 0.176576343768 -1.069847285211
H 1.181847251974 -0.007144125197 -2.930712574839
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000141159893 -0.000195076933 0.000029768603
C -0.000183982515 0.000096067279 -0.000262932323
O -0.000001796177 -0.000054734447 -0.000096552237
H 0.000003135318 0.000031109720 0.000031863200
H 0.000008987594 0.000023401289 -0.000006122490
H 0.000043242108 0.000031141158 0.000144813204
H 0.000049109192 0.000056398126 0.000073929087
C -0.000353206083 -0.000220826336 0.000215764840
O 0.000238978278 0.000171721409 -0.000042216781
H -0.000006163421 0.000006294027 -0.000023701013
H 0.000078943964 0.000051735301 -0.000097337133
*** Time spent in gradient calculation: 1.89 sec ***
* Info * Energy : -268.1267051717 a.u.
* Info * Gradient : 2.232823e-04 a.u. (RMS)
* Info * 4.691197e-04 a.u. (Max)
* Info * Time : 4.17 sec
Optimization Step 188
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.900450738908 0.986238124597 1.143005553729
C -0.856998757849 0.356205142072 1.699580227582
O -0.072996264280 -0.587476334084 1.138063033874
H -2.451080895434 1.733668762539 1.715814435134
H -2.232405901587 0.770713196242 0.123198333200
H -0.538307925515 0.575451531326 2.725139158031
H -0.362623231187 -0.764748720242 0.220519363900
C 0.841340065066 -0.189251142021 -1.888048188643
O -0.209415084965 -0.731709516563 -1.648045872455
H 1.515853000484 0.170725587545 -1.076119665057
H 1.186497045740 -0.023131321941 -2.935910967752
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000079458931 -0.000130730066 -0.000117175857
C -0.000071992444 0.000151169255 0.000220837153
O -0.000165367879 0.000063229097 -0.000285408329
H 0.000105018124 -0.000010706509 0.000025156903
H -0.000010118766 -0.000022638990 0.000051775841
H -0.000043977676 -0.000043838196 -0.000079913435
H 0.000092442227 -0.000054656795 0.000186492960
C 0.000135790111 -0.000001807895 -0.000112116645
O 0.000051044758 0.000083011400 0.000004001063
H -0.000080583039 -0.000006708980 -0.000013945416
H -0.000073049519 -0.000030756102 0.000088474582
*** Time spent in gradient calculation: 1.88 sec ***
* Info * Energy : -268.1267054705 a.u.
* Info * Gradient : 1.808402e-04 a.u. (RMS)
* Info * 3.358606e-04 a.u. (Max)
* Info * Time : 4.08 sec
Optimization Step 189
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.901802728332 0.980674703622 1.144682913314
C -0.856554558058 0.353230852014 1.701633915724
O -0.068268601266 -0.586923415238 1.140282468484
H -2.457975093184 1.724203194020 1.717273725698
H -2.230716079381 0.765474857800 0.123663955944
H -0.539839508780 0.572761833101 2.727845634918
H -0.357217059472 -0.764613694756 0.222120240496
C 0.840642636702 -0.185626339903 -1.888784867738
O -0.206162140099 -0.734716722067 -1.645944446204
H 1.516547170083 0.176683807156 -1.078829739704
H 1.181338077126 -0.015972986821 -2.937691532685
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000131227120 0.000183645545 0.000025897384
C 0.000163531717 -0.000157948293 0.000066042296
O 0.000072227270 -0.000002582107 0.000183125575
H -0.000046969282 -0.000016443803 -0.000035825741
H 0.000002954324 -0.000010461509 -0.000024016919
H -0.000001262079 0.000008384823 -0.000055166338
H -0.000080655705 -0.000016082016 -0.000152396989
C 0.000160971834 0.000142691271 -0.000098204162
O -0.000147083954 -0.000115846139 0.000023658867
H 0.000040777746 -0.000003701783 0.000022103734
H -0.000015385991 -0.000015656683 0.000012411986
*** Time spent in gradient calculation: 1.93 sec ***
* Info * Energy : -268.1267055916 a.u.
* Info * Gradient : 1.590648e-04 a.u. (RMS)
* Info * 2.367528e-04 a.u. (Max)
* Info * Time : 4.22 sec
Optimization Step 190
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.901620300770 0.979821906844 1.143649494903
C -0.855787288846 0.353651365408 1.700445146608
O -0.067158871382 -0.586366526788 1.139246204051
H -2.457153458303 1.723811219701 1.716308607407
H -2.231219496110 0.764027191998 0.123040334997
H -0.538768727871 0.573848538905 2.726507731482
H -0.356341684511 -0.764732555998 0.221484207702
C 0.840017658728 -0.185595232637 -1.887941201815
O -0.207292157751 -0.734088355276 -1.646614634425
H 1.514771597641 0.176550620810 -1.077035115441
H 1.182475287095 -0.015966121502 -2.936359626232
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000012904444 -0.000005405648 0.000003971059
C -0.000016474665 -0.000009120383 -0.000017012795
O -0.000001931584 -0.000004207324 0.000012281055
H -0.000001732755 0.000003513428 -0.000002649732
H -0.000000784028 -0.000000431740 0.000001771437
H 0.000006632724 0.000000773599 0.000007897964
H 0.000008494443 0.000001285934 -0.000013193247
C -0.000037813615 -0.000005578686 0.000016325153
O 0.000030936994 0.000014047842 -0.000021229224
H 0.000006773700 -0.000001401262 -0.000007628836
H 0.000011258090 0.000002753198 -0.000012718326
*** Time spent in gradient calculation: 2.04 sec ***
* Info * Energy : -268.1267059850 a.u.
* Info * Gradient : 2.162427e-05 a.u. (RMS)
* Info * 4.156323e-05 a.u. (Max)
* Info * Time : 4.23 sec
Optimization Step 191
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.900603502897 0.981819944134 1.141798476306
C -0.854657711870 0.356342014095 1.699235603254
O -0.067411169775 -0.585881981115 1.139822580710
H -2.455231616046 1.727720039179 1.712875509512
H -2.231372956091 0.763442638104 0.122099835652
H -0.536382503491 0.579234098824 2.724368345857
H -0.357808629192 -0.765973915967 0.222728220437
C 0.839316910378 -0.187174586504 -1.886536786411
O -0.209192278496 -0.733812199377 -1.646136259274
H 1.514165496144 0.173271661187 -1.074974791190
H 1.182809562935 -0.017757122808 -2.934688824844
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000003781722 0.000019984175 0.000018577911
C 0.000033337569 -0.000075304207 -0.000080717640
O 0.000026690429 0.000017632480 0.000095371577
H -0.000027330498 0.000007747499 -0.000010477008
H -0.000005388004 0.000001409931 -0.000006665597
H 0.000011957976 0.000017422055 0.000029098359
H -0.000034377799 -0.000021544641 -0.000046262047
C -0.000016193751 0.000010556202 0.000027425047
O 0.000007532928 0.000025890481 -0.000020279432
H -0.000001662575 -0.000004643454 -0.000006583035
H 0.000020044753 -0.000002683572 -0.000031950108
*** Time spent in gradient calculation: 2.07 sec ***
* Info * Energy : -268.1267056490 a.u.
* Info * Gradient : 5.560288e-05 a.u. (RMS)
* Info * 1.153146e-04 a.u. (Max)
* Info * Time : 4.45 sec
Optimization Step 192
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.900628266774 0.979847662687 1.141858342080
C -0.854439717315 0.354681454460 1.699162289685
O -0.066587430024 -0.586665242057 1.139078794938
H -2.455371365771 1.725091842093 1.713594824699
H -2.230853881906 0.762272880197 0.121835305792
H -0.536695567857 0.576738145804 2.724509442942
H -0.356796105551 -0.766554749992 0.222027918002
C 0.839181374071 -0.185818105583 -1.886525380970
O -0.208260001466 -0.734144630145 -1.645062729093
H 1.514290769977 0.175327119484 -1.075497738877
H 1.180768110661 -0.015160708356 -2.934942431669
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000033911457 -0.000020942481 -0.000030605062
C 0.000037164036 0.000033238649 0.000064789335
O -0.000025134962 -0.000012649971 -0.000113017012
H 0.000028309738 -0.000013371368 0.000005176610
H 0.000009683963 0.000001050020 -0.000008021358
H -0.000015608803 -0.000013254001 -0.000039274828
H -0.000002464827 -0.000006063898 0.000067451472
C 0.000109720035 0.000013628089 -0.000077973180
O -0.000037085694 0.000013472935 0.000055875606
H -0.000020756649 0.000003313922 0.000011826624
H -0.000031917283 -0.000001804458 0.000031311053
*** Time spent in gradient calculation: 1.91 sec ***
* Info * Energy : -268.1267057943 a.u.
* Info * Gradient : 7.138955e-05 a.u. (RMS)
* Info * 1.352924e-04 a.u. (Max)
* Info * Time : 4.14 sec
Optimization Step 193
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.900761552248 0.980935726369 1.143104891607
C -0.854943164732 0.355137076618 1.700313167263
O -0.066862836283 -0.585812762171 1.139878002192
H -2.455687959173 1.725871730479 1.715089931730
H -2.230887051462 0.763850072746 0.122953878760
H -0.537445072637 0.576436007889 2.725946000813
H -0.356086547954 -0.764880917088 0.222292695080
C 0.839225754780 -0.186526012834 -1.887544694277
O -0.208028711920 -0.734917585063 -1.645749067905
H 1.514439931522 0.175308346572 -1.076885271373
H 1.181041734889 -0.016675884195 -2.936084216126
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000014494828 -0.000012978022 -0.000015607102
C -0.000022664000 0.000020524866 0.000040604503
O -0.000033022076 -0.000002178312 -0.000029200047
H 0.000016089571 -0.000003783580 0.000000336840
H 0.000000669604 -0.000007676623 0.000007992253
H -0.000000558530 -0.000005849564 -0.000015574076
H 0.000036189663 -0.000000317794 0.000000700750
C -0.000027314052 -0.000006423077 -0.000009050171
O 0.000038211067 0.000020654724 -0.000022024576
H 0.000000506893 -0.000004625330 -0.000001030130
H -0.000004385092 -0.000000930377 0.000010546533
*** Time spent in gradient calculation: 1.90 sec ***
* Info * Energy : -268.1267058866 a.u.
* Info * Gradient : 3.094360e-05 a.u. (RMS)
* Info * 5.082964e-05 a.u. (Max)
* Info * Time : 3.85 sec
Optimization Step 194
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.899861121037 0.979836160204 1.141932572897
C -0.854074809615 0.354238065201 1.699507692104
O -0.066106295965 -0.587216922012 1.139780624172
H -2.454992417499 1.725093812841 1.713340328451
H -2.229910379686 0.762252583174 0.121837636317
H -0.536548567198 0.576273670632 2.725029122977
H -0.355991847234 -0.767014741573 0.222484457078
C 0.838535452346 -0.185998513078 -1.886908504603
O -0.208242268335 -0.735706068800 -1.645909912113
H 1.512953843804 0.176243012636 -1.075773097110
H 1.180799231187 -0.015365466269 -2.935236011489
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C 0.000003987246 0.000009606763 0.000015159031
C 0.000009353948 -0.000037678581 -0.000049216888
O 0.000023451464 -0.000000332167 0.000057057354
H -0.000014622451 0.000004882479 -0.000008566863
H -0.000001631418 0.000001352650 -0.000005598711
H 0.000008350431 0.000005439376 0.000013773696
H -0.000016093669 -0.000002584923 -0.000032979769
C -0.000015052802 0.000014616120 0.000024038945
O -0.000003864304 0.000002127214 -0.000021039130
H 0.000009309078 -0.000001888583 -0.000007184630
H 0.000015010179 0.000001087204 -0.000017857306
*** Time spent in gradient calculation: 1.99 sec ***
* Info * Energy : -268.1267057810 a.u.
* Info * Gradient : 3.334360e-05 a.u. (RMS)
* Info * 6.268552e-05 a.u. (Max)
* Info * Time : 4.06 sec
Optimization Step 195
=======================
* Info * Computing energy and gradient...
SCF Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -1.900173440343 0.980344909623 1.142484657510
C -0.854297427849 0.354747973981 1.699879904097
O -0.066286170440 -0.586477485561 1.139829807643
H -2.455204179530 1.725450768664 1.714165249788
H -2.230214822372 0.763031881842 0.122337228444
H -0.536780203183 0.576506743551 2.725427196022
H -0.356150650610 -0.766168206810 0.222556514082
C 0.838802869366 -0.186475488202 -1.887296470569
O -0.208110140013 -0.735577602069 -1.645474336000
H 1.513580390698 0.176157091282 -1.076640908641
H 1.180640953194 -0.016713915476 -2.935862055640
Analytical Gradient (Hartree/Bohr)
------------------------------------
Atom Gradient X Gradient Y Gradient Z
C -0.000003755863 -0.000003373913 -0.000001735717
C 0.000009767474 -0.000009465533 -0.000000287725
O 0.000000452757 -0.000004197698 -0.000008534879
H 0.000002042989 -0.000000484424 -0.000003481283
H 0.000001681571 0.000000672553 -0.000004245110
H 0.000000095010 -0.000001499612 -0.000005282332
H -0.000004182433 -0.000003168776 0.000004579314
C 0.000022480934 0.000010293362 -0.000012936807
O -0.000006520434 0.000007522938 0.000002103652
H -0.000002917314 -0.000000065103 -0.000001374867
H -0.000000935193 0.000000315889 -0.000001256062
*** Time spent in gradient calculation: 1.86 sec ***
* Info * Energy : -268.1267058432 a.u.
* Info * Gradient : 1.093763e-05 a.u. (RMS)
* Info * 2.790532e-05 a.u. (Max)
* Info * Time : 3.70 sec
* Info * Geometry optimization completed.
Final Geometry (Angstroms)
============================
Atom Coordinate X Coordinate Y Coordinate Z
C -1.900173440343 0.980344909623 1.142484657510
C -0.854297427849 0.354747973981 1.699879904097
O -0.066286170440 -0.586477485561 1.139829807643
H -2.455204179530 1.725450768664 1.714165249788
H -2.230214822372 0.763031881842 0.122337228444
H -0.536780203183 0.576506743551 2.725427196022
H -0.356150650610 -0.766168206810 0.222556514082
C 0.838802869366 -0.186475488202 -1.887296470569
O -0.208110140013 -0.735577602069 -1.645474336000
H 1.513580390698 0.176157091282 -1.076640908641
H 1.180640953194 -0.016713915476 -2.935862055640
Summary of IRC Calculation
============================
IRC Pt. Energy (a.u.) Energy Change (a.u.) E - E(TS) (kJ/mol) Displacement (RMS, Max)
-------------------------------------------------------------------------------------------------------------------
0 -268.159574501426 0.000000000000 -151.112 0.000e+00 0.000e+00
1 -268.159574546635 -0.000000045209 -151.112 4.855e-04 1.068e-03
2 -268.159574379447 0.000000167189 -151.112 9.452e-04 2.166e-03
3 -268.159571523422 0.000002856025 -151.104 1.676e-03 3.307e-03
4 -268.159573201466 -0.000001678044 -151.109 1.363e-03 2.825e-03
5 -268.159570046253 0.000003155213 -151.101 2.859e-03 6.615e-03
6 -268.159569523100 0.000000523153 -151.099 2.145e-03 3.860e-03
7 -268.159567126706 0.000002396394 -151.093 4.685e-03 1.035e-02
8 -268.159555941987 0.000011184719 -151.063 8.980e-03 2.074e-02
9 -268.159533050145 0.000022891842 -151.003 1.033e-02 2.288e-02
10 -268.159476151039 0.000056899107 -150.854 1.640e-02 3.515e-02
11 -268.159385902174 0.000090248865 -150.617 1.779e-02 3.903e-02
12 -268.159262172943 0.000123729231 -150.292 1.899e-02 4.184e-02
13 -268.159102822887 0.000159350056 -149.874 2.021e-02 4.535e-02
14 -268.158940032615 0.000162790272 -149.446 1.756e-02 3.793e-02
15 -268.158778530429 0.000161502186 -149.022 1.541e-02 3.311e-02
16 -268.158616097857 0.000162432572 -148.596 1.446e-02 3.070e-02
17 -268.158437579366 0.000178518491 -148.127 1.544e-02 3.351e-02
18 -268.158225120173 0.000212459194 -147.569 1.747e-02 3.796e-02
19 -268.157960318967 0.000264801206 -146.874 2.107e-02 4.678e-02
20 -268.157683693608 0.000276625358 -146.148 2.113e-02 4.734e-02
21 -268.157400881858 0.000282811751 -145.405 2.150e-02 4.799e-02
22 -268.157122669914 0.000278211943 -144.675 2.076e-02 4.583e-02
23 -268.156837147301 0.000285522614 -143.925 1.956e-02 4.214e-02
24 -268.156527420560 0.000309726741 -143.112 1.967e-02 4.145e-02
25 -268.156250462089 0.000276958471 -142.385 1.452e-02 3.069e-02
26 -268.156015469214 0.000234992875 -141.768 1.167e-02 2.451e-02
27 -268.155829094184 0.000186375029 -141.279 7.796e-03 1.636e-02
28 -268.155686378735 0.000142715450 -140.904 5.632e-03 1.174e-02
29 -268.155578009048 0.000108369686 -140.619 3.961e-03 7.898e-03
30 -268.155486895932 0.000091113116 -140.380 3.182e-03 6.281e-03
31 -268.155389865853 0.000097030078 -140.125 2.561e-03 5.208e-03
32 -268.155057855163 0.000332010690 -139.254 1.613e-02 2.954e-02
33 -268.154643840903 0.000414014260 -138.167 1.452e-02 2.740e-02
34 -268.154076494677 0.000567346226 -136.677 1.518e-02 2.383e-02
35 -268.153329977048 0.000746517629 -134.717 2.089e-02 3.669e-02
36 -268.152319828445 0.001010148603 -132.065 1.629e-02 3.318e-02
37 -268.151277671181 0.001042157264 -129.329 1.443e-02 2.723e-02
38 -268.149837106513 0.001440564668 -125.547 1.687e-02 2.760e-02
39 -268.147356037189 0.002481069324 -119.033 2.002e-02 3.381e-02
40 -268.143850219335 0.003505817854 -109.828 1.873e-02 3.346e-02
41 -268.139626380858 0.004223838477 -98.738 1.791e-02 3.215e-02
42 -268.135011892293 0.004614488565 -86.623 1.665e-02 2.719e-02
43 -268.130940938972 0.004070953320 -75.935 1.493e-02 2.473e-02
44 -268.126166188200 0.004774750772 -63.399 1.862e-02 3.196e-02
45 -268.121605142170 0.004561046030 -51.424 1.698e-02 3.197e-02
46 -268.116723059704 0.004882082467 -38.606 1.787e-02 3.107e-02
47 -268.111961629194 0.004761430510 -26.105 2.047e-02 4.830e-02
48 -268.107070947649 0.004890681545 -13.264 3.210e-02 9.069e-02
49 -268.102904985717 0.004165961932 -2.326 3.187e-02 9.094e-02
50 -268.102018893678 0.000886092038 0.000 2.750e-02 7.518e-02 <- TS
51 -268.102036078258 -0.000017184580 -0.045 2.681e-02 7.330e-02
52 -268.102039246622 -0.000003168364 -0.053 3.499e-05 1.032e-04
53 -268.102043698556 -0.000004451934 -0.065 4.945e-05 1.458e-04
54 -268.102049937111 -0.000006238555 -0.082 6.986e-05 2.059e-04
55 -268.102058645566 -0.000008708456 -0.104 9.858e-05 2.904e-04
56 -268.102070734322 -0.000012088756 -0.136 1.391e-04 4.096e-04
57 -268.102087381241 -0.000016646919 -0.180 1.960e-04 5.767e-04
58 -268.102110038442 -0.000022657201 -0.239 2.756e-04 8.102e-04
59 -268.102140349251 -0.000030310809 -0.319 3.863e-04 1.134e-03
60 -268.102179867820 -0.000039518569 -0.423 5.383e-04 1.574e-03
61 -268.102229427951 -0.000049560131 -0.553 7.405e-04 2.152e-03
62 -268.102288074610 -0.000058646658 -0.707 9.879e-04 2.828e-03
63 -268.102352528902 -0.000064454292 -0.876 1.214e-03 3.314e-03
64 -268.102422033440 -0.000069504538 -1.058 1.284e-03 2.874e-03
65 -268.102509421679 -0.000087388239 -1.288 1.487e-03 2.393e-03
66 -268.102634080073 -0.000124658394 -1.615 2.163e-03 3.513e-03
67 -268.102822817386 -0.000188737312 -2.111 3.607e-03 7.926e-03
68 -268.103113846568 -0.000291029183 -2.875 4.515e-03 8.191e-03
69 -268.103549990295 -0.000436143727 -4.020 7.347e-03 1.659e-02
70 -268.103560344610 -0.000010354315 -4.047 1.461e-04 3.205e-04
71 -268.103574877042 -0.000014532433 -4.085 1.856e-04 3.453e-04
72 -268.103595158067 -0.000020281025 -4.138 2.555e-04 4.459e-04
73 -268.103623508320 -0.000028350253 -4.213 3.545e-04 5.845e-04
74 -268.103663300537 -0.000039792216 -4.317 4.992e-04 8.075e-04
75 -268.103719504198 -0.000056203661 -4.465 7.125e-04 1.170e-03
76 -268.103728896660 -0.000009392462 -4.490 1.240e-04 2.214e-04
77 -268.103742225823 -0.000013329163 -4.525 1.716e-04 2.929e-04
78 -268.103761106082 -0.000018880259 -4.574 2.441e-04 4.220e-04
79 -268.103787873808 -0.000026767726 -4.644 3.467e-04 6.048e-04
80 -268.103825861386 -0.000037987578 -4.744 4.930e-04 8.690e-04
81 -268.103879865954 -0.000054004567 -4.886 7.005e-04 1.247e-03
82 -268.103956796562 -0.000076930609 -5.088 9.943e-04 1.781e-03
83 -268.104066684140 -0.000109887577 -5.376 1.409e-03 2.528e-03
84 -268.104224143518 -0.000157459378 -5.790 1.991e-03 3.562e-03
85 -268.104450545849 -0.000226402331 -6.384 2.810e-03 4.975e-03
86 -268.104777090693 -0.000326544844 -7.242 3.957e-03 6.879e-03
87 -268.105248803197 -0.000471712504 -8.480 5.561e-03 9.376e-03
88 -268.105928658528 -0.000679855332 -10.265 7.835e-03 1.270e-02
89 -268.106888309727 -0.000959651198 -12.785 1.081e-02 1.749e-02
90 -268.108138748072 -0.001250438346 -16.068 1.525e-02 2.653e-02
91 -268.109353890122 -0.001215142050 -19.258 1.518e-02 2.455e-02
92 -268.110317958183 -0.000964068061 -21.789 1.436e-02 2.661e-02
93 -268.111203916368 -0.000885958184 -24.115 1.194e-02 1.937e-02
94 -268.112112157358 -0.000908240990 -26.500 1.399e-02 2.119e-02
95 -268.112979986257 -0.000867828899 -28.778 1.359e-02 2.240e-02
96 -268.113812664405 -0.000832678148 -30.965 1.509e-02 2.351e-02
97 -268.114608415251 -0.000795750846 -33.054 1.517e-02 2.466e-02
98 -268.115393114449 -0.000784699198 -35.114 1.667e-02 2.630e-02
99 -268.116031658865 -0.000638544416 -36.791 1.848e-02 2.856e-02
100 -268.116611873256 -0.000580214390 -38.314 1.496e-02 2.350e-02
101 -268.116997876508 -0.000386003252 -39.327 1.354e-02 2.145e-02
102 -268.117319511218 -0.000321634710 -40.172 9.481e-03 1.553e-02
103 -268.117698392383 -0.000378881165 -41.167 1.213e-02 1.747e-02
104 -268.118149487326 -0.000451094943 -42.351 1.661e-02 2.636e-02
105 -268.118538045119 -0.000388557793 -43.371 1.734e-02 2.571e-02
106 -268.118761118274 -0.000223073155 -43.957 9.638e-03 1.543e-02
107 -268.119006553822 -0.000245435548 -44.601 1.162e-02 1.839e-02
108 -268.119272612219 -0.000266058397 -45.300 1.413e-02 2.299e-02
109 -268.119568727332 -0.000296115113 -46.077 1.727e-02 3.077e-02
110 -268.119873797840 -0.000305070508 -46.878 2.052e-02 3.502e-02
111 -268.120129921924 -0.000256124085 -47.550 1.818e-02 3.735e-02
112 -268.120324769351 -0.000194847427 -48.062 1.451e-02 2.791e-02
113 -268.120567869971 -0.000243100619 -48.700 1.901e-02 3.914e-02
114 -268.120877317821 -0.000309447851 -49.513 2.531e-02 5.679e-02
115 -268.121080309586 -0.000202991764 -50.046 1.767e-02 4.173e-02
116 -268.121194085401 -0.000113775815 -50.344 9.775e-03 2.277e-02
117 -268.121292718170 -0.000098632769 -50.603 8.413e-03 1.977e-02
118 -268.121401513562 -0.000108795392 -50.889 8.665e-03 2.039e-02
119 -268.121548695086 -0.000147181524 -51.275 1.172e-02 2.840e-02
120 -268.121784983890 -0.000236288804 -51.896 1.974e-02 5.108e-02
121 -268.121940769865 -0.000155785975 -52.305 1.236e-02 2.943e-02
122 -268.122165570534 -0.000224800670 -52.895 1.794e-02 4.396e-02
123 -268.122453838468 -0.000288267933 -53.652 2.314e-02 5.782e-02
124 -268.122685884743 -0.000232046276 -54.261 1.854e-02 4.682e-02
125 -268.122990639631 -0.000304754887 -55.061 2.417e-02 5.990e-02
126 -268.123322726196 -0.000332086566 -55.933 2.766e-02 6.762e-02
127 -268.123521073207 -0.000198347011 -56.454 1.499e-02 3.692e-02
128 -268.123857603610 -0.000336530402 -57.338 2.776e-02 6.814e-02
129 -268.124108653008 -0.000251049398 -57.997 2.126e-02 5.249e-02
130 -268.124421360668 -0.000312707660 -58.818 2.603e-02 6.225e-02
131 -268.124718240936 -0.000296880268 -59.597 2.768e-02 6.653e-02
132 -268.124980187079 -0.000261946143 -60.285 2.525e-02 5.883e-02
133 -268.125203790332 -0.000223603252 -60.872 2.464e-02 5.730e-02
134 -268.125382733487 -0.000178943155 -61.342 2.082e-02 4.640e-02
135 -268.125523440566 -0.000140707079 -61.711 1.858e-02 4.098e-02
136 -268.125641798847 -0.000118358282 -62.022 1.658e-02 3.468e-02
137 -268.125743492179 -0.000101693332 -62.289 1.597e-02 3.264e-02
138 -268.125836757973 -0.000093265793 -62.534 1.592e-02 3.009e-02
139 -268.125921303442 -0.000084545469 -62.756 1.642e-02 3.037e-02
140 -268.125997331779 -0.000076028337 -62.955 1.671e-02 2.679e-02
141 -268.126049435416 -0.000052103637 -63.092 1.279e-02 2.079e-02
142 -268.126095275410 -0.000045839994 -63.213 1.257e-02 1.953e-02
143 -268.126137035162 -0.000041759753 -63.322 1.274e-02 2.003e-02
144 -268.126180663221 -0.000043628059 -63.437 1.500e-02 2.327e-02
145 -268.126221666191 -0.000041002970 -63.544 1.600e-02 2.607e-02
146 -268.126262522798 -0.000040856607 -63.652 1.828e-02 3.221e-02
147 -268.126298372449 -0.000035849651 -63.746 1.813e-02 3.383e-02
148 -268.126328727065 -0.000030354616 -63.825 1.773e-02 3.373e-02
149 -268.126349375822 -0.000020648757 -63.880 1.134e-02 2.542e-02
150 -268.126380629721 -0.000031253899 -63.962 1.802e-02 3.844e-02
151 -268.126402335932 -0.000021706211 -64.019 1.289e-02 3.030e-02
152 -268.126423248855 -0.000020912924 -64.074 1.103e-02 2.340e-02
153 -268.126446475750 -0.000023226895 -64.135 1.312e-02 2.989e-02
154 -268.126466067401 -0.000019591651 -64.186 1.090e-02 2.576e-02
155 -268.126489781194 -0.000023713794 -64.248 1.342e-02 2.994e-02
156 -268.126512867808 -0.000023086614 -64.309 1.294e-02 2.706e-02
157 -268.126534905841 -0.000022038033 -64.367 1.331e-02 2.868e-02
158 -268.126556421116 -0.000021515276 -64.423 1.305e-02 2.584e-02
159 -268.126576294989 -0.000019873873 -64.475 1.290e-02 2.431e-02
160 -268.126593270618 -0.000016975629 -64.520 1.178e-02 2.294e-02
161 -268.126607935201 -0.000014664583 -64.559 1.079e-02 1.833e-02
162 -268.126621491946 -0.000013556744 -64.594 1.086e-02 1.935e-02
163 -268.126633755924 -0.000012263979 -64.626 1.065e-02 1.871e-02
164 -268.126644369052 -0.000010613128 -64.654 9.876e-03 1.536e-02
165 -268.126653982402 -0.000009613350 -64.679 9.864e-03 1.733e-02
166 -268.126663068811 -0.000009086408 -64.703 1.002e-02 1.496e-02
167 -268.126671255161 -0.000008186350 -64.725 1.029e-02 1.699e-02
168 -268.126678955296 -0.000007700135 -64.745 1.065e-02 1.528e-02
169 -268.126684660886 -0.000005705590 -64.760 9.853e-03 1.624e-02
170 -268.126688039154 -0.000003378268 -64.769 5.692e-03 8.128e-03
171 -268.126690893319 -0.000002854165 -64.776 5.822e-03 9.303e-03
172 -268.126693882703 -0.000002989383 -64.784 6.463e-03 9.103e-03
173 -268.126696654922 -0.000002772219 -64.791 7.322e-03 1.054e-02
174 -268.126698980187 -0.000002325266 -64.798 6.752e-03 9.612e-03
175 -268.126700278115 -0.000001297927 -64.801 4.274e-03 5.892e-03
176 -268.126701479315 -0.000001201201 -64.804 4.286e-03 6.347e-03
177 -268.126702661271 -0.000001181956 -64.807 4.890e-03 7.016e-03
178 -268.126703860054 -0.000001198782 -64.810 5.737e-03 8.057e-03
179 -268.126704609531 -0.000000749477 -64.812 5.578e-03 8.456e-03
180 -268.126580123763 0.000124485768 -64.486 1.027e-01 1.719e-01
181 -268.126681035993 -0.000100912230 -64.750 5.190e-02 8.882e-02
182 -268.126701641142 -0.000020605149 -64.805 2.580e-02 4.297e-02
183 -268.126705171687 -0.000003530545 -64.814 1.270e-02 2.079e-02
184 -268.126705470472 -0.000000298785 -64.815 6.377e-03 1.106e-02
185 -268.126705591556 -0.000000121084 -64.815 2.844e-03 5.045e-03
186 -268.126705984986 -0.000000393430 -64.816 1.536e-03 2.919e-03
187 -268.126705648993 0.000000335993 -64.815 2.110e-03 3.243e-03
188 -268.126705794278 -0.000000145285 -64.815 9.995e-04 1.901e-03
189 -268.126705886630 -0.000000092352 -64.816 1.410e-03 1.878e-03
190 -268.126705781040 0.000000105590 -64.815 1.500e-03 2.397e-03
191 -268.126705843209 -0.000000062169 -64.816 7.538e-04 1.166e-03
IRC Path Summary:
Transition State at point 50
Forward barrier (TS - Reactant): 151.112 kJ/mol
Backward barrier (TS - Product): 64.816 kJ/mol
Reaction energy (Product - Reactant): 86.297 kJ/mol
*** Time spent in Optimization Driver: 889.59 sec
* Info * Optimization results written to file: vlx_20260417_2f17ea33.h5
opt_drv.plot_irc(opt_results)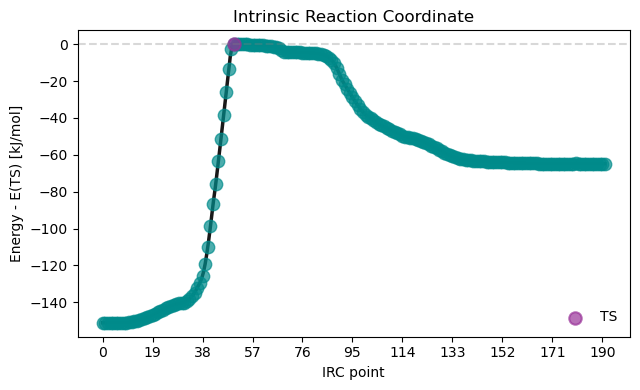
Text file
@jobs
task: optimize
@end
@method settings
xcfun: b3lyp
basis: def2-svp
dispersion: yes
@end
@optimize
irc: yes
@end
@molecule
charge: -1
multiplicity: 1
xyz:
...
@endExcited state optimization¶
Python script
import veloxchem as vlx
molecule = vlx.Molecule.read_name("bithiophene")
basis = vlx.MolecularBasis.read(molecule, "def2-svp")
scf_drv = vlx.ScfRestrictedDriver()
scf_drv.xcfun = "b3lyp"
scf_results = scf_drv.compute(molecule, basis)
rsp_drv = vlx.LinearResponseEigenSolver()
rsp_drv.nstates = 2
rsp_results = rsp_drv.compute(molecule, basis, scf_results)
grad_drv = vlx.TddftGradientDriver(scf_drv)
grad_drv.state_deriv_index = 1
opt_drv = vlx.OptimizationDriver(grad_drv)
opt_results = opt_drv.compute(molecule, basis, scf_drv, rsp_drv, rsp_results)
Reading bithiophene from PubChem...
Reference: S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang, E. E. Bolton, Nucleic Acids Res., 2025, 53, D1516-D1525.
Please double-check the compound since names may refer to more than one record.
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Kohn-Sham
Initial Guess Model : Superposition of Atomic Densities
Convergence Accelerator : Two Level Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Starting Reduced Basis SCF calculation...
* Info * ...done. SCF energy in reduced basis set: -1100.725787840755 a.u. Time: 0.90 sec.
Iter. | Kohn-Sham Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -1104.338004312942 0.0000000000 0.49202145 0.02656548 0.00000000
2 -1104.350259865760 -0.0122555528 0.39397503 0.02213500 0.44278499
3 -1104.370526045550 -0.0202661798 0.09781896 0.00613034 0.20954288
4 -1104.371648437448 -0.0011223919 0.03405146 0.00252953 0.05036528
5 -1104.371780461838 -0.0001320244 0.01363532 0.00097332 0.01779208
6 -1104.371807106211 -0.0000266444 0.00176156 0.00009070 0.00694527
7 -1104.371807514470 -0.0000004083 0.00090795 0.00005092 0.00152418
8 -1104.371807623264 -0.0000001088 0.00014010 0.00000754 0.00037102
9 -1104.371807625891 -0.0000000026 0.00006435 0.00000313 0.00008385
10 -1104.371807626426 -0.0000000005 0.00001779 0.00000069 0.00002970
11 -1104.371807626474 -0.0000000000 0.00000232 0.00000011 0.00001062
12 -1104.371807626475 -0.0000000000 0.00000035 0.00000002 0.00000179
*** SCF converged in 12 iterations. Time: 9.17 sec.
Spin-Restricted Kohn-Sham:
--------------------------
Total Energy : -1104.3718076265 a.u.
Electronic Energy : -1740.3108753445 a.u.
Nuclear Repulsion Energy : 635.9390677180 a.u.
------------------------------------
Gradient Norm : 0.0000003526 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
Linear Response EigenSolver Setup
===================================
Number of States : 2
Max. Number of Iterations : 150
Convergence Threshold : 1.0e-04
ERI Screening Threshold : 1.0e-12
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * 6 gerade trial vectors in reduced space
* Info * 6 ungerade trial vectors in reduced space
*** Iteration: 1 * Residuals (Max,Min): 3.53e-01 and 1.52e-01
* Info * 8 gerade trial vectors in reduced space
* Info * 8 ungerade trial vectors in reduced space
*** Iteration: 2 * Residuals (Max,Min): 7.82e-02 and 5.77e-02
* Info * 10 gerade trial vectors in reduced space
* Info * 10 ungerade trial vectors in reduced space
*** Iteration: 3 * Residuals (Max,Min): 3.04e-02 and 2.36e-02
* Info * 12 gerade trial vectors in reduced space
* Info * 12 ungerade trial vectors in reduced space
*** Iteration: 4 * Residuals (Max,Min): 9.21e-03 and 7.08e-03
* Info * 14 gerade trial vectors in reduced space
* Info * 14 ungerade trial vectors in reduced space
*** Iteration: 5 * Residuals (Max,Min): 3.48e-03 and 3.02e-03
* Info * 16 gerade trial vectors in reduced space
* Info * 16 ungerade trial vectors in reduced space
*** Iteration: 6 * Residuals (Max,Min): 1.15e-03 and 1.14e-03
* Info * 18 gerade trial vectors in reduced space
* Info * 18 ungerade trial vectors in reduced space
*** Iteration: 7 * Residuals (Max,Min): 3.04e-04 and 2.78e-04
* Info * 20 gerade trial vectors in reduced space
* Info * 20 ungerade trial vectors in reduced space
*** Iteration: 8 * Residuals (Max,Min): 8.30e-05 and 7.45e-05
*** Linear response converged in 8 iterations. Time: 15.71 sec
Electric Transition Dipole Moments (dipole length, a.u.)
--------------------------------------------------------
X Y Z
Excited State S1: -1.933610 -0.783237 -0.625102
Excited State S2: -0.000011 0.000032 -0.000016
Electric Transition Dipole Moments (dipole velocity, a.u.)
----------------------------------------------------------
X Y Z
Excited State S1: -1.947054 -0.776311 -0.632695
Excited State S2: -0.000010 0.000029 -0.000016
Magnetic Transition Dipole Moments (a.u.)
-----------------------------------------
X Y Z
Excited State S1: -0.047876 0.067531 0.064469
Excited State S2: -0.096091 0.058738 0.223648
One-Photon Absorption
---------------------
Excited State S1: 0.15519545 a.u. 4.22308 eV Osc.Str. 0.4907
Excited State S2: 0.18551106 a.u. 5.04801 eV Osc.Str. 0.0000
Electronic Circular Dichroism
-----------------------------
Excited State S1: Rot.Str. 0.000004 a.u. 0.0020 [10**(-40) cgs]
Excited State S2: Rot.Str. -0.000001 a.u. -0.0004 [10**(-40) cgs]
Character of excitations:
Excited state 1
---------------
HOMO -> LUMO -0.9952
Excited state 2
---------------
HOMO-1 -> LUMO 0.9010
HOMO -> LUMO+1 -0.3823
Optimization Driver Setup
===========================
Coordinate System : TRIC
Constraints : No
Max. Number of Steps : 300
Transition State : No
IRC : No
Hessian : never
* Info * Using geomeTRIC for geometry optimization.
L.-P. Wang and C.C. Song, J. Chem. Phys. 2016, 144, 214108
Optimization Step 0
=====================
* Info * Computing energy and gradient...
RPA Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Excited States of Interest : 1
* Info * Computing orbital response...
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Computing the right-hand side (RHS) of CPHF/CPKS equations...
* Info * RHS of CPHF/CPKS equations computed in 2.80 sec.
Coupled-Perturbed Kohn-Sham Solver Setup
------------------------------------------
Solver Type : Iterative Subspace Algorithm
Max. Number of Iterations : 150
Convergence Threshold : 1.0e-04
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * 1 trial vectors in reduced space
*** Iteration: 1 * Residuals (Max,Min): 3.27e-01 and 3.27e-01
* Info * 2 trial vectors in reduced space
*** Iteration: 2 * Residuals (Max,Min): 9.88e-02 and 9.88e-02
* Info * 3 trial vectors in reduced space
*** Iteration: 3 * Residuals (Max,Min): 3.53e-02 and 3.53e-02
* Info * 4 trial vectors in reduced space
*** Iteration: 4 * Residuals (Max,Min): 7.05e-03 and 7.05e-03
* Info * 5 trial vectors in reduced space
*** Iteration: 5 * Residuals (Max,Min): 2.76e-03 and 2.76e-03
* Info * 6 trial vectors in reduced space
*** Iteration: 6 * Residuals (Max,Min): 6.86e-04 and 6.86e-04
* Info * 7 trial vectors in reduced space
*** Iteration: 7 * Residuals (Max,Min): 1.84e-04 and 1.84e-04
* Info * 8 trial vectors in reduced space
*** Iteration: 8 * Residuals (Max,Min): 4.96e-05 and 4.96e-05
*** Coupled-Perturbed Kohn-Sham converged in 8 iterations. Time: 9.89 sec
* Info * Computing the omega Lagrange multipliers...
* Info * The omega Lagrange multipliers computed in 1.16 sec.
* Info * Computing ground-state gradient...
* Info * Computing excited-state gradient...
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.456943000000 0.250181000000 -1.298614000000
C -2.434044000000 -1.090326000000 -0.936664000000
S -0.951126000000 -1.415293000000 -0.214158000000
C -0.328315000000 0.142071000000 -0.355598000000
C -1.282628000000 0.941060000000 -0.975504000000
C 1.000572000000 0.577542000000 0.101047000000
C 1.954886000000 -0.221457000000 0.720936000000
C 3.129208000000 0.469416000000 1.044060000000
C 3.106308000000 1.809932000000 0.682131000000
S 1.623402000000 2.134896000000 -0.040401000000
H -3.303202000000 0.716832000000 -1.784808000000
H -3.234074000000 -1.800918000000 -1.093817000000
H -1.140467000000 1.992011000000 -1.190436000000
H 1.812696000000 -1.272405000000 0.935866000000
H 3.975469000000 0.002762000000 1.530254000000
H 3.906347000000 2.520525000000 0.839260000000
Analytical Gradient (Hartree/Bohr)
------------------------------------
Excited State 1
Atom Gradient X Gradient Y Gradient Z
C -0.002374051528 -0.020418273570 0.004342529461
C 0.012210964184 -0.015225840549 0.009249034160
S -0.023482871218 0.080422084741 -0.031214529458
C -0.064337443042 -0.048688987622 -0.014859037877
C 0.033730853685 -0.023115994987 0.020565535101
C 0.064336216836 0.048687931869 0.014859763260
C -0.033733324292 0.023115175638 -0.020568053734
C 0.002376492892 0.020417860710 -0.004342600224
C -0.012212207851 0.015228193918 -0.009247600015
S 0.023483450237 -0.080424223801 0.031214766394
H 0.005597858627 -0.002869514333 0.003158591629
H -0.000372219162 0.007866301884 -0.002227002279
H -0.002220680705 -0.006282038857 0.000696616250
H 0.002218687237 0.006281940249 -0.000696888497
H -0.005595840059 0.002869273550 -0.003157573122
H 0.000374121353 -0.007863888921 0.002226446749
*** Time spent in gradient calculation: 40.34 sec ***
* Info * Energy : -1104.2166121745 a.u.
* Info * Gradient : 4.728735e-02 a.u. (RMS)
* Info * 8.940856e-02 a.u. (Max)
* Info * Time : 66.60 sec
Optimization Step 1
=====================
* Info * Computing energy and gradient...
RPA Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Excited States of Interest : 1
* Info * Computing orbital response...
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Computing the right-hand side (RHS) of CPHF/CPKS equations...
* Info * RHS of CPHF/CPKS equations computed in 2.60 sec.
Coupled-Perturbed Kohn-Sham Solver Setup
------------------------------------------
Solver Type : Iterative Subspace Algorithm
Max. Number of Iterations : 150
Convergence Threshold : 1.0e-04
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * 1 trial vectors in reduced space
*** Iteration: 1 * Residuals (Max,Min): 3.18e-01 and 3.18e-01
* Info * 2 trial vectors in reduced space
*** Iteration: 2 * Residuals (Max,Min): 7.54e-02 and 7.54e-02
* Info * 3 trial vectors in reduced space
*** Iteration: 3 * Residuals (Max,Min): 3.25e-02 and 3.25e-02
* Info * 4 trial vectors in reduced space
*** Iteration: 4 * Residuals (Max,Min): 5.77e-03 and 5.77e-03
* Info * 5 trial vectors in reduced space
*** Iteration: 5 * Residuals (Max,Min): 2.86e-03 and 2.86e-03
* Info * 6 trial vectors in reduced space
*** Iteration: 6 * Residuals (Max,Min): 7.28e-04 and 7.28e-04
* Info * 7 trial vectors in reduced space
*** Iteration: 7 * Residuals (Max,Min): 2.55e-04 and 2.55e-04
* Info * 8 trial vectors in reduced space
*** Iteration: 8 * Residuals (Max,Min): 8.78e-05 and 8.78e-05
*** Coupled-Perturbed Kohn-Sham converged in 8 iterations. Time: 8.83 sec
* Info * Computing the omega Lagrange multipliers...
* Info * The omega Lagrange multipliers computed in 0.87 sec.
* Info * Computing ground-state gradient...
* Info * Computing excited-state gradient...
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.430896297798 0.302095173326 -1.301063994141
C -2.313607029302 -1.043825387247 -0.897139371039
S -0.838920524177 -1.487164977602 -0.147064139211
C -0.317726480996 0.154533697228 -0.354322874428
C -1.271066842820 1.014559023387 -0.989847198473
C 0.961730623581 0.565930243237 0.087406639619
C 1.935663849296 -0.271717228162 0.725885823836
C 3.107107413441 0.412404921960 1.049546180273
C 2.999869087807 1.757239364081 0.650240001847
S 1.525645845698 2.199776462470 -0.099449049022
H -3.307696844885 0.727063795208 -1.789430340551
H -3.100408261998 -1.779367776499 -1.042052425864
H -1.115705275706 2.072181705147 -1.200860907420
H 1.779100585649 -1.329266987958 0.936377227561
H 3.976087182623 -0.027661741039 1.538520315613
H 3.788911569253 2.490048717006 0.796808233800
Analytical Gradient (Hartree/Bohr)
------------------------------------
Excited State 1
Atom Gradient X Gradient Y Gradient Z
C -0.012654511935 0.005959632571 -0.007002837922
C 0.030099348272 0.000543908720 0.012792267002
S 0.003767294651 0.009079618428 -0.000765743702
C -0.034862654298 -0.039008880331 -0.004737096349
C 0.010601108825 0.008220445570 0.002396097323
C 0.019347231763 0.032525157245 -0.000224933391
C -0.005030311293 0.003757322971 -0.003150677837
C 0.015845823818 -0.009088032743 0.009195008684
C -0.027993207656 -0.000340201422 -0.011938816307
S -0.000276387783 -0.012588604499 0.003186433878
H 0.001428726118 0.002551280998 -0.000056506930
H 0.000544005484 0.004358931765 -0.000912229028
H -0.003392490379 -0.000001316956 -0.001456720248
H 0.003478968917 0.000994081082 0.001233538560
H -0.000790220003 -0.002085810771 0.000208757502
H -0.000103440727 -0.004871249240 0.001235168983
*** Time spent in gradient calculation: 37.02 sec ***
* Info * Energy : -1104.2377719182 a.u.
* Info * Gradient : 2.156252e-02 a.u. (RMS)
* Info * 5.253130e-02 a.u. (Max)
* Info * Time : 62.88 sec
Optimization Step 2
=====================
* Info * Computing energy and gradient...
RPA Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Excited States of Interest : 1
* Info * Computing orbital response...
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Computing the right-hand side (RHS) of CPHF/CPKS equations...
* Info * RHS of CPHF/CPKS equations computed in 2.56 sec.
Coupled-Perturbed Kohn-Sham Solver Setup
------------------------------------------
Solver Type : Iterative Subspace Algorithm
Max. Number of Iterations : 150
Convergence Threshold : 1.0e-04
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * 1 trial vectors in reduced space
*** Iteration: 1 * Residuals (Max,Min): 3.34e-01 and 3.34e-01
* Info * 2 trial vectors in reduced space
*** Iteration: 2 * Residuals (Max,Min): 1.07e-01 and 1.07e-01
* Info * 3 trial vectors in reduced space
*** Iteration: 3 * Residuals (Max,Min): 4.04e-02 and 4.04e-02
* Info * 4 trial vectors in reduced space
*** Iteration: 4 * Residuals (Max,Min): 5.88e-03 and 5.88e-03
* Info * 5 trial vectors in reduced space
*** Iteration: 5 * Residuals (Max,Min): 2.97e-03 and 2.97e-03
* Info * 6 trial vectors in reduced space
*** Iteration: 6 * Residuals (Max,Min): 9.34e-04 and 9.34e-04
* Info * 7 trial vectors in reduced space
*** Iteration: 7 * Residuals (Max,Min): 3.18e-04 and 3.18e-04
* Info * 8 trial vectors in reduced space
*** Iteration: 8 * Residuals (Max,Min): 9.70e-05 and 9.70e-05
*** Coupled-Perturbed Kohn-Sham converged in 8 iterations. Time: 9.55 sec
* Info * Computing the omega Lagrange multipliers...
* Info * The omega Lagrange multipliers computed in 1.22 sec.
* Info * Computing ground-state gradient...
* Info * Computing excited-state gradient...
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.434127331455 0.298685957487 -1.301565545756
C -2.422312203520 -1.064322352439 -0.938482796008
S -0.915649451442 -1.503435074883 -0.175766634799
C -0.283922597909 0.160095597917 -0.341258392530
C -1.270849041902 1.002750474114 -0.986660484127
C 0.977039486711 0.501616722463 0.110874283739
C 1.976911365347 -0.326439001847 0.758009100555
C 3.099761568515 0.452466342674 1.035890523949
C 3.061150100307 1.811089290833 0.662445510628
S 1.539236656387 2.189089956946 -0.090780681700
H -3.299657728443 0.752153302851 -1.792565556524
H -3.218027298035 -1.798712070741 -1.087513153313
H -1.101104752673 2.062856007196 -1.192152915414
H 1.847286509070 -1.389843813159 0.981619688384
H 3.984856197984 0.037796529353 1.525111127885
H 3.836181994427 2.570091238167 0.796107921500
Analytical Gradient (Hartree/Bohr)
------------------------------------
Excited State 1
Atom Gradient X Gradient Y Gradient Z
C 0.004451373752 0.005772272262 0.000396641527
C -0.006178765086 -0.012366958463 0.000595567260
S 0.002291686171 0.004087270976 -0.000084803579
C 0.017376447352 -0.005756301118 0.008976026722
C -0.003919965698 0.009011535623 -0.004052361270
C -0.007663147996 0.010172616468 -0.005964094552
C 0.005724816021 -0.022450062256 0.008356984813
C -0.016417059948 0.002052415261 -0.007594314474
C 0.009224484251 0.013333385893 0.000462992031
S -0.002587029272 -0.003045399311 -0.000312071089
H -0.000780539148 0.000334063107 -0.000422950124
H -0.002734225645 -0.001136286305 -0.000876775460
H -0.000150347506 0.001868463108 -0.000554998319
H -0.000095701175 -0.003793954664 0.000955308467
H -0.000803716639 -0.000441392583 -0.000229511335
H 0.002248323748 0.002360204021 0.000346242420
*** Time spent in gradient calculation: 39.43 sec ***
* Info * Energy : -1104.2435302178 a.u.
* Info * Gradient : 1.186258e-02 a.u. (RMS)
* Info * 2.462962e-02 a.u. (Max)
* Info * Time : 64.07 sec
Optimization Step 3
=====================
* Info * Computing energy and gradient...
RPA Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Excited States of Interest : 1
* Info * Computing orbital response...
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Computing the right-hand side (RHS) of CPHF/CPKS equations...
* Info * RHS of CPHF/CPKS equations computed in 2.38 sec.
Coupled-Perturbed Kohn-Sham Solver Setup
------------------------------------------
Solver Type : Iterative Subspace Algorithm
Max. Number of Iterations : 150
Convergence Threshold : 1.0e-04
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * 1 trial vectors in reduced space
*** Iteration: 1 * Residuals (Max,Min): 3.18e-01 and 3.18e-01
* Info * 2 trial vectors in reduced space
*** Iteration: 2 * Residuals (Max,Min): 8.07e-02 and 8.07e-02
* Info * 3 trial vectors in reduced space
*** Iteration: 3 * Residuals (Max,Min): 3.39e-02 and 3.39e-02
* Info * 4 trial vectors in reduced space
*** Iteration: 4 * Residuals (Max,Min): 6.43e-03 and 6.43e-03
* Info * 5 trial vectors in reduced space
*** Iteration: 5 * Residuals (Max,Min): 3.14e-03 and 3.14e-03
* Info * 6 trial vectors in reduced space
*** Iteration: 6 * Residuals (Max,Min): 1.07e-03 and 1.07e-03
* Info * 7 trial vectors in reduced space
*** Iteration: 7 * Residuals (Max,Min): 4.12e-04 and 4.12e-04
* Info * 8 trial vectors in reduced space
*** Iteration: 8 * Residuals (Max,Min): 1.25e-04 and 1.25e-04
* Info * 9 trial vectors in reduced space
*** Iteration: 9 * Residuals (Max,Min): 4.10e-05 and 4.10e-05
*** Coupled-Perturbed Kohn-Sham converged in 9 iterations. Time: 9.71 sec
* Info * Computing the omega Lagrange multipliers...
* Info * The omega Lagrange multipliers computed in 0.92 sec.
* Info * Computing ground-state gradient...
* Info * Computing excited-state gradient...
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.475146919215 0.296983149740 -1.318779979259
C -2.462165145512 -1.055631609157 -0.957996069150
S -0.957953919046 -1.502399415438 -0.194354935420
C -0.295713365385 0.157909568153 -0.345777462842
C -1.298293577094 0.969166774350 -0.989634756279
C 0.976623649159 0.491597214794 0.113313010772
C 2.019714264908 -0.263436038460 0.759873708897
C 3.181905367773 0.442765511143 1.073744268728
C 3.070578068172 1.781806700676 0.674137447863
S 1.550380945343 2.179106315416 -0.083408762519
H -3.335115276516 0.758586277192 -1.809500307590
H -3.251638538807 -1.794498446771 -1.103186710858
H -1.119800944238 2.026566558957 -1.190614146321
H 1.880314135818 -1.322758954104 0.978251783487
H 4.068511021323 0.030470920710 1.563011926718
H 3.825818699877 2.559237672890 0.794420906359
Analytical Gradient (Hartree/Bohr)
------------------------------------
Excited State 1
Atom Gradient X Gradient Y Gradient Z
C -0.001361554620 0.004080741918 -0.001656152065
C -0.006582636014 -0.003405186360 -0.001932547526
S -0.000435756360 0.007957623111 -0.002274609308
C 0.014299983942 0.006788844718 0.004362572095
C -0.007107939264 -0.008940840217 -0.000707214119
C -0.011764324868 -0.005320768799 -0.003659221202
C 0.007762454758 0.016870694119 -0.001095206985
C 0.013708464922 -0.007749535733 0.007926785148
C -0.003736631417 -0.006396231116 0.000075215788
S -0.006015171868 -0.003029617590 -0.001788916085
H -0.001360623293 -0.000837460097 -0.000364371509
H -0.001462829022 -0.000106754417 -0.000600621465
H 0.000263783033 -0.000788902941 0.000320270171
H 0.000547751506 0.001648601869 -0.000197939511
H 0.002557354780 -0.000027273806 0.001106096554
H 0.000670558061 -0.000743211787 0.000483515493
*** Time spent in gradient calculation: 39.29 sec ***
* Info * Energy : -1104.2433636325 a.u.
* Info * Gradient : 9.707104e-03 a.u. (RMS)
* Info * 1.860310e-02 a.u. (Max)
* Info * Time : 66.64 sec
Optimization Step 4
=====================
* Info * Computing energy and gradient...
RPA Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Excited States of Interest : 1
* Info * Computing orbital response...
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Computing the right-hand side (RHS) of CPHF/CPKS equations...
* Info * RHS of CPHF/CPKS equations computed in 2.60 sec.
Coupled-Perturbed Kohn-Sham Solver Setup
------------------------------------------
Solver Type : Iterative Subspace Algorithm
Max. Number of Iterations : 150
Convergence Threshold : 1.0e-04
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * 1 trial vectors in reduced space
*** Iteration: 1 * Residuals (Max,Min): 3.21e-01 and 3.21e-01
* Info * 2 trial vectors in reduced space
*** Iteration: 2 * Residuals (Max,Min): 8.10e-02 and 8.10e-02
* Info * 3 trial vectors in reduced space
*** Iteration: 3 * Residuals (Max,Min): 3.53e-02 and 3.53e-02
* Info * 4 trial vectors in reduced space
*** Iteration: 4 * Residuals (Max,Min): 6.22e-03 and 6.22e-03
* Info * 5 trial vectors in reduced space
*** Iteration: 5 * Residuals (Max,Min): 3.10e-03 and 3.10e-03
* Info * 6 trial vectors in reduced space
*** Iteration: 6 * Residuals (Max,Min): 9.80e-04 and 9.80e-04
* Info * 7 trial vectors in reduced space
*** Iteration: 7 * Residuals (Max,Min): 3.70e-04 and 3.70e-04
* Info * 8 trial vectors in reduced space
*** Iteration: 8 * Residuals (Max,Min): 1.15e-04 and 1.15e-04
* Info * 9 trial vectors in reduced space
*** Iteration: 9 * Residuals (Max,Min): 3.96e-05 and 3.96e-05
*** Coupled-Perturbed Kohn-Sham converged in 9 iterations. Time: 9.83 sec
* Info * Computing the omega Lagrange multipliers...
* Info * The omega Lagrange multipliers computed in 0.97 sec.
* Info * Computing ground-state gradient...
* Info * Computing excited-state gradient...
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.461082169685 0.292941196382 -1.311679389899
C -2.433742434795 -1.057966156838 -0.945176600772
S -0.931642421987 -1.502068710164 -0.183143384207
C -0.297759122296 0.166814442386 -0.348999134939
C -1.292954290074 0.983432552608 -0.991089956402
C 0.973336798254 0.509101415184 0.107300645092
C 1.993059183517 -0.272099616522 0.750694255349
C 3.152350753584 0.438990819082 1.062034078668
C 3.070696898453 1.783468748468 0.673748925687
S 1.558486130984 2.194881876756 -0.084072018472
H -3.326906674074 0.742878725985 -1.801851599068
H -3.218288973639 -1.801591605811 -1.087001114265
H -1.119774788540 2.041378224920 -1.194493762407
H 1.843500575185 -1.331299203316 0.964675476152
H 4.033078308197 0.017046423657 1.551310947200
H 3.835843587946 2.549556270811 0.801268196407
Analytical Gradient (Hartree/Bohr)
------------------------------------
Excited State 1
Atom Gradient X Gradient Y Gradient Z
C 0.000210201248 0.002514186441 -0.000568926685
C -0.003861999323 -0.001974710683 -0.001139708179
S 0.000266593567 0.004067506641 -0.000952015082
C 0.007843679442 0.001932189076 0.002863814732
C -0.004665819940 -0.003563625094 -0.001069875793
C -0.006987183672 -0.002230180361 -0.002417867188
C 0.004675320005 0.008343468857 -0.000181891249
C 0.006493343473 -0.004919003684 0.004083182733
C -0.001325629975 -0.002301572561 0.000034625375
S -0.003321942999 -0.001642685124 -0.000996229302
H -0.000695128987 -0.000531373652 -0.000158833362
H -0.000732303455 -0.000029866711 -0.000306911962
H 0.000169081762 -0.000215769023 0.000129036302
H 0.000183004449 0.000728561045 -0.000113010286
H 0.001278531954 0.000121455447 0.000517534011
H 0.000458538335 -0.000296321569 0.000274927569
*** Time spent in gradient calculation: 39.11 sec ***
* Info * Energy : -1104.2455853168 a.u.
* Info * Gradient : 5.085141e-03 a.u. (RMS)
* Info * 9.565834e-03 a.u. (Max)
* Info * Time : 65.29 sec
Optimization Step 5
=====================
* Info * Computing energy and gradient...
RPA Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Excited States of Interest : 1
* Info * Computing orbital response...
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Computing the right-hand side (RHS) of CPHF/CPKS equations...
* Info * RHS of CPHF/CPKS equations computed in 2.78 sec.
Coupled-Perturbed Kohn-Sham Solver Setup
------------------------------------------
Solver Type : Iterative Subspace Algorithm
Max. Number of Iterations : 150
Convergence Threshold : 1.0e-04
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * 1 trial vectors in reduced space
*** Iteration: 1 * Residuals (Max,Min): 3.23e-01 and 3.23e-01
* Info * 2 trial vectors in reduced space
*** Iteration: 2 * Residuals (Max,Min): 8.32e-02 and 8.32e-02
* Info * 3 trial vectors in reduced space
*** Iteration: 3 * Residuals (Max,Min): 3.65e-02 and 3.65e-02
* Info * 4 trial vectors in reduced space
*** Iteration: 4 * Residuals (Max,Min): 5.90e-03 and 5.90e-03
* Info * 5 trial vectors in reduced space
*** Iteration: 5 * Residuals (Max,Min): 2.95e-03 and 2.95e-03
* Info * 6 trial vectors in reduced space
*** Iteration: 6 * Residuals (Max,Min): 8.78e-04 and 8.78e-04
* Info * 7 trial vectors in reduced space
*** Iteration: 7 * Residuals (Max,Min): 2.62e-04 and 2.62e-04
* Info * 8 trial vectors in reduced space
*** Iteration: 8 * Residuals (Max,Min): 7.61e-05 and 7.61e-05
*** Coupled-Perturbed Kohn-Sham converged in 8 iterations. Time: 9.47 sec
* Info * Computing the omega Lagrange multipliers...
* Info * The omega Lagrange multipliers computed in 1.00 sec.
* Info * Computing ground-state gradient...
* Info * Computing excited-state gradient...
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.451688281560 0.287366329538 -1.306247896860
C -2.400567242132 -1.059487614107 -0.930589343578
S -0.900252816489 -1.496094915356 -0.171277178377
C -0.297204425992 0.182187761089 -0.352819341083
C -1.289876919484 0.993688475228 -0.992490330869
C 0.973689629820 0.534870149570 0.100687614585
C 1.962355810038 -0.279104430615 0.739340050320
C 3.120872377827 0.433812575923 1.049861679337
C 3.072286756889 1.781860615146 0.674870490749
S 1.573457206080 2.217638625300 -0.083584485823
H -3.323701133262 0.724752841736 -1.795797954277
H -3.176672191501 -1.811878101862 -1.066486935967
H -1.124799286428 2.051798470302 -1.199402177698
H 1.798823099376 -1.337539768771 0.947112819924
H 3.993417601652 -0.002178741399 1.539299882505
H 3.849373510444 2.533124959871 0.811411442424
Analytical Gradient (Hartree/Bohr)
------------------------------------
Excited State 1
Atom Gradient X Gradient Y Gradient Z
C -0.001144893185 0.000805353635 -0.000703627392
C 0.000460617936 0.001007224339 -0.000065551669
S 0.000576281279 0.000864635684 0.000020301778
C 0.001172845563 -0.001976719652 0.001023936314
C -0.000897053646 -0.001433545258 -0.000008924071
C 0.001307886542 0.001348466661 0.000207226832
C -0.000202081925 -0.000722574221 0.000104295599
C -0.000797885266 -0.000124422464 -0.000310180097
C -0.000255234254 0.000201768127 -0.000164138144
S 0.000227879392 0.000289901481 0.000021284295
H -0.000218764938 -0.000090992996 -0.000069903108
H 0.000065439046 0.000201286738 -0.000024538183
H -0.000004338469 -0.000293484786 0.000074991052
H -0.000090445791 -0.000081963383 -0.000017359676
H -0.000100535910 0.000064605961 -0.000060167247
H -0.000098117322 -0.000057734828 -0.000027178016
*** Time spent in gradient calculation: 38.13 sec ***
* Info * Energy : -1104.2463016517 a.u.
* Info * Gradient : 1.100143e-03 a.u. (RMS)
* Info * 2.516234e-03 a.u. (Max)
* Info * Time : 62.45 sec
Optimization Step 6
=====================
* Info * Computing energy and gradient...
RPA Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Excited States of Interest : 1
* Info * Computing orbital response...
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Computing the right-hand side (RHS) of CPHF/CPKS equations...
* Info * RHS of CPHF/CPKS equations computed in 2.52 sec.
Coupled-Perturbed Kohn-Sham Solver Setup
------------------------------------------
Solver Type : Iterative Subspace Algorithm
Max. Number of Iterations : 150
Convergence Threshold : 1.0e-04
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * 1 trial vectors in reduced space
*** Iteration: 1 * Residuals (Max,Min): 3.25e-01 and 3.25e-01
* Info * 2 trial vectors in reduced space
*** Iteration: 2 * Residuals (Max,Min): 8.75e-02 and 8.75e-02
* Info * 3 trial vectors in reduced space
*** Iteration: 3 * Residuals (Max,Min): 3.77e-02 and 3.77e-02
* Info * 4 trial vectors in reduced space
*** Iteration: 4 * Residuals (Max,Min): 5.92e-03 and 5.92e-03
* Info * 5 trial vectors in reduced space
*** Iteration: 5 * Residuals (Max,Min): 2.96e-03 and 2.96e-03
* Info * 6 trial vectors in reduced space
*** Iteration: 6 * Residuals (Max,Min): 9.15e-04 and 9.15e-04
* Info * 7 trial vectors in reduced space
*** Iteration: 7 * Residuals (Max,Min): 2.92e-04 and 2.92e-04
* Info * 8 trial vectors in reduced space
*** Iteration: 8 * Residuals (Max,Min): 9.51e-05 and 9.51e-05
*** Coupled-Perturbed Kohn-Sham converged in 8 iterations. Time: 8.78 sec
* Info * Computing the omega Lagrange multipliers...
* Info * The omega Lagrange multipliers computed in 0.92 sec.
* Info * Computing ground-state gradient...
* Info * Computing excited-state gradient...
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.441205856191 0.284513621543 -1.301033956428
C -2.402190093194 -1.064516829141 -0.929996551937
S -0.901822266266 -1.497649665345 -0.171557191111
C -0.303164509008 0.184212354312 -0.355933417225
C -1.286371285878 1.005998054332 -0.994249584794
C 0.968540459435 0.529878295488 0.099782683878
C 1.961569867627 -0.278548247816 0.738845196220
C 3.121319171682 0.433053624660 1.050272597696
C 3.069863922151 1.780302241864 0.674292101389
S 1.569487568515 2.212796877824 -0.083981202645
H -3.313238089728 0.721368589517 -1.790462939535
H -3.180105265041 -1.815085760443 -1.067143869961
H -1.123031873896 2.064618673068 -1.202045652214
H 1.800254991928 -1.337034126287 0.947564421900
H 3.994025810946 -0.002861379359 1.539760164802
H 3.845489840975 2.533419140916 0.809739785316
Analytical Gradient (Hartree/Bohr)
------------------------------------
Excited State 1
Atom Gradient X Gradient Y Gradient Z
C 0.004105463695 -0.002057870530 0.002304250263
C -0.002058604035 -0.001555056393 -0.000475175304
S -0.000012202052 0.000313880785 -0.000087682230
C -0.003052595119 -0.001734667358 -0.000856224310
C 0.000594399312 0.004935936089 -0.001040806772
C -0.000765594912 -0.000113028736 -0.000299588025
C 0.000226881288 0.000928252715 -0.000145565572
C 0.000438980063 -0.000367801187 0.000284767400
C -0.000179897562 -0.000244112146 -0.000013631115
S -0.000115969177 -0.000240014706 0.000012507902
H 0.000484574488 -0.000111463899 0.000237674869
H 0.000050727168 -0.000400576309 0.000127459072
H 0.000284144708 0.000511604920 -0.000012321577
H -0.000044203652 0.000132604435 -0.000053542379
H 0.000076532523 0.000069169836 0.000014680584
H -0.000033177118 -0.000066437003 0.000002881824
*** Time spent in gradient calculation: 37.22 sec ***
* Info * Energy : -1104.2462142464 a.u.
* Info * Gradient : 2.169502e-03 a.u. (RMS)
* Info * 5.138018e-03 a.u. (Max)
* Info * Time : 60.40 sec
Optimization Step 7
=====================
* Info * Computing energy and gradient...
RPA Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Excited States of Interest : 1
* Info * Computing orbital response...
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Computing the right-hand side (RHS) of CPHF/CPKS equations...
* Info * RHS of CPHF/CPKS equations computed in 2.44 sec.
Coupled-Perturbed Kohn-Sham Solver Setup
------------------------------------------
Solver Type : Iterative Subspace Algorithm
Max. Number of Iterations : 150
Convergence Threshold : 1.0e-04
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * 1 trial vectors in reduced space
*** Iteration: 1 * Residuals (Max,Min): 3.24e-01 and 3.24e-01
* Info * 2 trial vectors in reduced space
*** Iteration: 2 * Residuals (Max,Min): 8.52e-02 and 8.52e-02
* Info * 3 trial vectors in reduced space
*** Iteration: 3 * Residuals (Max,Min): 3.71e-02 and 3.71e-02
* Info * 4 trial vectors in reduced space
*** Iteration: 4 * Residuals (Max,Min): 5.90e-03 and 5.90e-03
* Info * 5 trial vectors in reduced space
*** Iteration: 5 * Residuals (Max,Min): 2.95e-03 and 2.95e-03
* Info * 6 trial vectors in reduced space
*** Iteration: 6 * Residuals (Max,Min): 8.88e-04 and 8.88e-04
* Info * 7 trial vectors in reduced space
*** Iteration: 7 * Residuals (Max,Min): 2.69e-04 and 2.69e-04
* Info * 8 trial vectors in reduced space
*** Iteration: 8 * Residuals (Max,Min): 8.15e-05 and 8.15e-05
*** Coupled-Perturbed Kohn-Sham converged in 8 iterations. Time: 8.98 sec
* Info * Computing the omega Lagrange multipliers...
* Info * The omega Lagrange multipliers computed in 0.90 sec.
* Info * Computing ground-state gradient...
* Info * Computing excited-state gradient...
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.446830838266 0.286327955100 -1.303894969046
C -2.402034523941 -1.061851100102 -0.930606155617
S -0.902051384031 -1.497454848234 -0.171696924474
C -0.300353232921 0.182589884635 -0.354281295019
C -1.288005465544 0.999420330841 -0.993196269375
C 0.970973414756 0.531310962320 0.100455171420
C 1.962765830940 -0.278942277965 0.739473174618
C 3.121733521568 0.433915521213 1.050209135747
C 3.071039076624 1.781429834206 0.674456252193
S 1.570834440923 2.214253914203 -0.083816801145
H -3.318635254559 0.723801977767 -1.793379954244
H -3.179379177075 -1.813015507836 -1.067358985628
H -1.123313410035 2.057828046152 -1.200351541985
H 1.801028323076 -1.337433994931 0.948029297637
H 3.994662722340 -0.001518580069 1.539666718803
H 3.847046148357 2.534090046665 0.810170091304
Analytical Gradient (Hartree/Bohr)
------------------------------------
Excited State 1
Atom Gradient X Gradient Y Gradient Z
C 0.001273184340 -0.000436831266 0.000661613487
C -0.000693257386 -0.000384237547 -0.000195832812
S 0.000230373875 0.000546457395 -0.000044778650
C -0.000903782929 -0.001616949439 0.000036848996
C -0.000044332674 0.001566027907 -0.000430230043
C 0.000005545090 0.000526336871 -0.000136473406
C 0.000105426194 0.000152614715 0.000006500765
C -0.000071101166 -0.000206500469 0.000023610370
C -0.000113441391 -0.000022930529 -0.000043938495
S -0.000018942171 -0.000112713203 0.000020931947
H 0.000131771537 -0.000116141029 0.000087330527
H 0.000031307749 -0.000112282519 0.000043184251
H 0.000148150404 0.000150913536 0.000023817310
H -0.000067452529 0.000044574596 -0.000040687681
H 0.000018602135 0.000075550785 -0.000011849766
H -0.000032384759 -0.000052805826 -0.000000231306
*** Time spent in gradient calculation: 37.75 sec ***
* Info * Energy : -1104.2463226000 a.u.
* Info * Gradient : 7.852832e-04 a.u. (RMS)
* Info * 1.852757e-03 a.u. (Max)
* Info * Time : 61.62 sec
Optimization Step 8
=====================
* Info * Computing energy and gradient...
RPA Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Excited States of Interest : 1
* Info * Computing orbital response...
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Computing the right-hand side (RHS) of CPHF/CPKS equations...
* Info * RHS of CPHF/CPKS equations computed in 2.59 sec.
Coupled-Perturbed Kohn-Sham Solver Setup
------------------------------------------
Solver Type : Iterative Subspace Algorithm
Max. Number of Iterations : 150
Convergence Threshold : 1.0e-04
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * 1 trial vectors in reduced space
*** Iteration: 1 * Residuals (Max,Min): 3.24e-01 and 3.24e-01
* Info * 2 trial vectors in reduced space
*** Iteration: 2 * Residuals (Max,Min): 8.44e-02 and 8.44e-02
* Info * 3 trial vectors in reduced space
*** Iteration: 3 * Residuals (Max,Min): 3.69e-02 and 3.69e-02
* Info * 4 trial vectors in reduced space
*** Iteration: 4 * Residuals (Max,Min): 5.92e-03 and 5.92e-03
* Info * 5 trial vectors in reduced space
*** Iteration: 5 * Residuals (Max,Min): 2.95e-03 and 2.95e-03
* Info * 6 trial vectors in reduced space
*** Iteration: 6 * Residuals (Max,Min): 8.88e-04 and 8.88e-04
* Info * 7 trial vectors in reduced space
*** Iteration: 7 * Residuals (Max,Min): 2.62e-04 and 2.62e-04
* Info * 8 trial vectors in reduced space
*** Iteration: 8 * Residuals (Max,Min): 7.25e-05 and 7.25e-05
*** Coupled-Perturbed Kohn-Sham converged in 8 iterations. Time: 9.20 sec
* Info * Computing the omega Lagrange multipliers...
* Info * The omega Lagrange multipliers computed in 0.99 sec.
* Info * Computing ground-state gradient...
* Info * Computing excited-state gradient...
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.450045030640 0.287794181666 -1.305694271790
C -2.403180520034 -1.060125200935 -0.931579606282
S -0.904099452252 -1.498535971061 -0.172299411825
C -0.300122289369 0.181926865576 -0.354020813587
C -1.289135096329 0.996952232316 -0.993058084149
C 0.971366638333 0.528575470073 0.101344426585
C 1.964912716279 -0.279825755011 0.740621133931
C 3.123195552794 0.434901634058 1.050597503448
C 3.072420657246 1.782409811791 0.674839521872
S 1.570844583602 2.212179181718 -0.083231676479
H -3.321234474365 0.726710789577 -1.795305011647
H -3.180630286412 -1.811191400926 -1.068401978243
H -1.124108223286 2.055312674512 -1.200060124023
H 1.805159380344 -1.338497711766 0.950059246808
H 3.996402836830 -0.000110607525 1.540062740164
H 3.847722128483 2.536079324398 0.810001573161
Analytical Gradient (Hartree/Bohr)
------------------------------------
Excited State 1
Atom Gradient X Gradient Y Gradient Z
C -0.000100432784 0.000182975835 -0.000091322395
C 0.000045709353 0.000206048595 -0.000033691946
S 0.000322706527 0.000174495052 0.000093177375
C -0.000145467970 -0.000586810820 0.000091790336
C -0.000191272569 0.000095387828 -0.000107289668
C -0.000433879205 -0.000089229532 -0.000163320003
C 0.000318240032 0.000079004067 0.000116620900
C -0.000213843907 -0.000014848765 -0.000088250178
C 0.000428608782 0.000219804825 0.000125955427
S -0.000076972228 -0.000382503892 0.000066797301
H -0.000045229581 -0.000015470647 -0.000015195101
H 0.000039716032 -0.000019887412 0.000022696156
H 0.000039353578 0.000022440758 0.000010927953
H -0.000066433358 0.000011115512 -0.000031218841
H 0.000032046796 0.000077975847 -0.000006741346
H 0.000045156517 0.000040651347 0.000008390038
*** Time spent in gradient calculation: 38.10 sec ***
* Info * Energy : -1104.2463329930 a.u.
* Info * Gradient : 3.059729e-04 a.u. (RMS)
* Info * 6.115009e-04 a.u. (Max)
* Info * Time : 62.45 sec
Optimization Step 9
=====================
* Info * Computing energy and gradient...
RPA Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Excited States of Interest : 1
* Info * Computing orbital response...
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Computing the right-hand side (RHS) of CPHF/CPKS equations...
* Info * RHS of CPHF/CPKS equations computed in 2.57 sec.
Coupled-Perturbed Kohn-Sham Solver Setup
------------------------------------------
Solver Type : Iterative Subspace Algorithm
Max. Number of Iterations : 150
Convergence Threshold : 1.0e-04
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * 1 trial vectors in reduced space
*** Iteration: 1 * Residuals (Max,Min): 3.24e-01 and 3.24e-01
* Info * 2 trial vectors in reduced space
*** Iteration: 2 * Residuals (Max,Min): 8.41e-02 and 8.41e-02
* Info * 3 trial vectors in reduced space
*** Iteration: 3 * Residuals (Max,Min): 3.68e-02 and 3.68e-02
* Info * 4 trial vectors in reduced space
*** Iteration: 4 * Residuals (Max,Min): 5.92e-03 and 5.92e-03
* Info * 5 trial vectors in reduced space
*** Iteration: 5 * Residuals (Max,Min): 2.95e-03 and 2.95e-03
* Info * 6 trial vectors in reduced space
*** Iteration: 6 * Residuals (Max,Min): 8.89e-04 and 8.89e-04
* Info * 7 trial vectors in reduced space
*** Iteration: 7 * Residuals (Max,Min): 2.64e-04 and 2.64e-04
* Info * 8 trial vectors in reduced space
*** Iteration: 8 * Residuals (Max,Min): 7.41e-05 and 7.41e-05
*** Coupled-Perturbed Kohn-Sham converged in 8 iterations. Time: 10.82 sec
* Info * Computing the omega Lagrange multipliers...
* Info * The omega Lagrange multipliers computed in 0.97 sec.
* Info * Computing ground-state gradient...
* Info * Computing excited-state gradient...
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.450165228084 0.286406417517 -1.305419391481
C -2.402306775030 -1.061266736736 -0.930944706870
S -0.902158373920 -1.496262165640 -0.172084783755
C -0.298849019888 0.185265475304 -0.354364004911
C -1.289955724625 0.997167560207 -0.993491169855
C 0.972458152475 0.532661631333 0.100744522162
C 1.963305749632 -0.279067110535 0.739730887867
C 3.122711733009 0.433341984125 1.050822521830
C 3.073131086548 1.781060490456 0.675542570029
S 1.573529983126 2.216046747444 -0.083057036607
H -3.321739705611 0.724576715241 -1.795010150094
H -3.179028323005 -1.813204661135 -1.067231137484
H -1.127274695801 2.055629589815 -1.201526696983
H 1.801535083127 -1.337641418649 0.948259457219
H 3.994912898043 -0.003620440722 1.540368457852
H 3.849650805475 2.533238904281 0.811633651888
Analytical Gradient (Hartree/Bohr)
------------------------------------
Excited State 1
Atom Gradient X Gradient Y Gradient Z
C -0.000036845366 0.000040026162 -0.000026257306
C -0.000259487697 0.000041149723 -0.000121942235
S 0.000115173728 0.000354429906 -0.000043131274
C 0.000448563678 -0.000355666130 0.000286364856
C -0.000207250886 -0.000147408724 -0.000050563488
C 0.000164304350 0.000228234309 0.000010645156
C -0.000022622109 -0.000153913402 0.000030788724
C 0.000022005484 -0.000072765764 0.000028461192
C -0.000159701336 -0.000060393117 -0.000053327580
S -0.000029620742 0.000253435787 -0.000079855822
H -0.000037478567 -0.000023947383 -0.000009692971
H -0.000007626656 -0.000010157385 -0.000000117796
H 0.000020077320 -0.000044574491 0.000020264592
H 0.000002095765 -0.000035282652 0.000010531892
H 0.000010689134 -0.000022014291 0.000010341721
H -0.000021996563 0.000009427123 -0.000012397186
*** Time spent in gradient calculation: 39.93 sec ***
* Info * Energy : -1104.2463352806 a.u.
* Info * Gradient : 2.407655e-04 a.u. (RMS)
* Info * 6.400880e-04 a.u. (Max)
* Info * Time : 64.29 sec
Optimization Step 10
======================
* Info * Computing energy and gradient...
RPA Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Excited States of Interest : 1
* Info * Computing orbital response...
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Computing the right-hand side (RHS) of CPHF/CPKS equations...
* Info * RHS of CPHF/CPKS equations computed in 2.45 sec.
Coupled-Perturbed Kohn-Sham Solver Setup
------------------------------------------
Solver Type : Iterative Subspace Algorithm
Max. Number of Iterations : 150
Convergence Threshold : 1.0e-04
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * 1 trial vectors in reduced space
*** Iteration: 1 * Residuals (Max,Min): 3.24e-01 and 3.24e-01
* Info * 2 trial vectors in reduced space
*** Iteration: 2 * Residuals (Max,Min): 8.42e-02 and 8.42e-02
* Info * 3 trial vectors in reduced space
*** Iteration: 3 * Residuals (Max,Min): 3.68e-02 and 3.68e-02
* Info * 4 trial vectors in reduced space
*** Iteration: 4 * Residuals (Max,Min): 5.92e-03 and 5.92e-03
* Info * 5 trial vectors in reduced space
*** Iteration: 5 * Residuals (Max,Min): 2.95e-03 and 2.95e-03
* Info * 6 trial vectors in reduced space
*** Iteration: 6 * Residuals (Max,Min): 8.90e-04 and 8.90e-04
* Info * 7 trial vectors in reduced space
*** Iteration: 7 * Residuals (Max,Min): 2.63e-04 and 2.63e-04
* Info * 8 trial vectors in reduced space
*** Iteration: 8 * Residuals (Max,Min): 7.19e-05 and 7.19e-05
*** Coupled-Perturbed Kohn-Sham converged in 8 iterations. Time: 9.25 sec
* Info * Computing the omega Lagrange multipliers...
* Info * The omega Lagrange multipliers computed in 1.02 sec.
* Info * Computing ground-state gradient...
* Info * Computing excited-state gradient...
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.449926981137 0.286492881680 -1.305364798421
C -2.401806392067 -1.061175817250 -0.930780312874
S -0.902075361737 -1.496601034361 -0.171979986269
C -0.299481207622 0.185474461750 -0.354700303321
C -1.289969756651 0.997817581486 -0.993683479217
C 0.971971709993 0.532269810447 0.100641781039
C 1.963034847115 -0.278975342908 0.739593911257
C 3.122561292303 0.433249567401 1.050802551075
C 3.073425113839 1.780930716412 0.675736705184
S 1.573657093358 2.215436392180 -0.082815741411
H -3.321568776464 0.724476213411 -1.794941452299
H -3.178599041003 -1.813002905026 -1.067131730338
H -1.127434019983 2.056306509621 -1.201787317365
H 1.801389921459 -1.337519164548 0.948155681363
H 3.994605263207 -0.003960882492 1.540348146758
H 3.850005845854 2.533024010391 0.811888622735
Analytical Gradient (Hartree/Bohr)
------------------------------------
Excited State 1
Atom Gradient X Gradient Y Gradient Z
C 0.000016107237 -0.000042242433 0.000018068124
C -0.000084215888 0.000084293032 -0.000057849325
S 0.000091300302 0.000136468776 0.000003753707
C -0.000008435697 -0.000267465082 0.000066851727
C -0.000048792716 0.000036774113 -0.000030852861
C 0.000045746526 0.000033929218 0.000010676494
C -0.000014775911 0.000006437255 -0.000007985907
C 0.000001177448 -0.000023084240 0.000006443299
C -0.000022432082 -0.000006750525 -0.000008389794
S 0.000012066461 0.000059056860 -0.000010830657
H -0.000002225854 -0.000005140734 0.000000476457
H 0.000016938274 -0.000009320243 0.000010244613
H 0.000008731378 -0.000009669330 0.000006201671
H -0.000003114950 0.000005578839 -0.000002415622
H 0.000001053731 0.000001214464 0.000000089290
H -0.000009168680 0.000000312652 -0.000004515609
*** Time spent in gradient calculation: 38.86 sec ***
* Info * Energy : -1104.2463366521 a.u.
* Info * Gradient : 9.258451e-05 a.u. (RMS)
* Info * 2.758222e-04 a.u. (Max)
* Info * Time : 64.41 sec
Optimization Step 11
======================
* Info * Computing energy and gradient...
RPA Gradient Driver Setup
===========================
Gradient Type : Analytical
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
Excited States of Interest : 1
* Info * Computing orbital response...
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Computing the right-hand side (RHS) of CPHF/CPKS equations...
* Info * RHS of CPHF/CPKS equations computed in 2.58 sec.
Coupled-Perturbed Kohn-Sham Solver Setup
------------------------------------------
Solver Type : Iterative Subspace Algorithm
Max. Number of Iterations : 150
Convergence Threshold : 1.0e-04
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * 1 trial vectors in reduced space
*** Iteration: 1 * Residuals (Max,Min): 3.24e-01 and 3.24e-01
* Info * 2 trial vectors in reduced space
*** Iteration: 2 * Residuals (Max,Min): 8.42e-02 and 8.42e-02
* Info * 3 trial vectors in reduced space
*** Iteration: 3 * Residuals (Max,Min): 3.68e-02 and 3.68e-02
* Info * 4 trial vectors in reduced space
*** Iteration: 4 * Residuals (Max,Min): 5.92e-03 and 5.92e-03
* Info * 5 trial vectors in reduced space
*** Iteration: 5 * Residuals (Max,Min): 2.95e-03 and 2.95e-03
* Info * 6 trial vectors in reduced space
*** Iteration: 6 * Residuals (Max,Min): 8.91e-04 and 8.91e-04
* Info * 7 trial vectors in reduced space
*** Iteration: 7 * Residuals (Max,Min): 2.63e-04 and 2.63e-04
* Info * 8 trial vectors in reduced space
*** Iteration: 8 * Residuals (Max,Min): 7.13e-05 and 7.13e-05
*** Coupled-Perturbed Kohn-Sham converged in 8 iterations. Time: 9.59 sec
* Info * Computing the omega Lagrange multipliers...
* Info * The omega Lagrange multipliers computed in 1.01 sec.
* Info * Computing ground-state gradient...
* Info * Computing excited-state gradient...
Molecular Geometry (Angstroms)
--------------------------------
Atom Coordinate X Coordinate Y Coordinate Z
C -2.449943398349 0.286593885193 -1.305439083621
C -2.401681849274 -1.061133308306 -0.930783713408
S -0.902074477791 -1.496481254924 -0.172042018505
C -0.299540304473 0.186058953814 -0.354894490688
C -1.290069879068 0.998154437206 -0.993839189567
C 0.971929709001 0.532334233831 0.100613342736
C 1.962876788537 -0.279093497186 0.739564621515
C 3.122513977555 0.432951828909 1.050896585261
C 3.073743937125 1.780659100918 0.676002462890
S 1.574038241582 2.215386072238 -0.082596273247
H -3.321616352183 0.724510560348 -1.795021001324
H -3.178545601317 -1.812896592891 -1.067193002622
H -1.127841980575 2.056678469874 -1.202081417709
H 1.801156299585 -1.337644426657 0.948074764873
H 3.994412560307 -0.004534330069 1.540456689580
H 3.850474249127 2.532590091349 0.812282191911
Analytical Gradient (Hartree/Bohr)
------------------------------------
Excited State 1
Atom Gradient X Gradient Y Gradient Z
C 0.000004148166 -0.000025171466 0.000008473614
C -0.000033437063 0.000028114716 -0.000021296851
S 0.000041361024 0.000037907309 0.000008208580
C -0.000017182809 -0.000077323105 0.000013119050
C 0.000004327926 0.000030833505 -0.000006480926
C -0.000018915336 -0.000038901485 0.000002034501
C -0.000005020110 0.000007576220 -0.000004172748
C -0.000005116249 0.000002168240 -0.000002921317
C 0.000009659940 0.000008220121 0.000001566022
S 0.000010831614 0.000026550003 -0.000002784808
H 0.000001825488 0.000002903131 0.000000073706
H 0.000007217598 -0.000009717052 0.000006209948
H 0.000000427941 0.000001825879 -0.000000417061
H 0.000001020901 -0.000000104250 0.000000877114
H -0.000000986466 0.000000695426 -0.000000670043
H -0.000000243874 0.000004610012 -0.000001857920
*** Time spent in gradient calculation: 38.51 sec ***
* Info * Energy : -1104.2463368307 a.u.
* Info * Gradient : 3.259065e-05 a.u. (RMS)
* Info * 8.028836e-05 a.u. (Max)
* Info * Time : 62.50 sec
* Info * Geometry optimization completed.
Final Geometry (Angstroms)
============================
Atom Coordinate X Coordinate Y Coordinate Z
C -2.449943398349 0.286593885193 -1.305439083621
C -2.401681849274 -1.061133308306 -0.930783713408
S -0.902074477791 -1.496481254924 -0.172042018505
C -0.299540304473 0.186058953814 -0.354894490688
C -1.290069879068 0.998154437206 -0.993839189567
C 0.971929709001 0.532334233831 0.100613342736
C 1.962876788537 -0.279093497186 0.739564621515
C 3.122513977555 0.432951828909 1.050896585261
C 3.073743937125 1.780659100918 0.676002462890
S 1.574038241582 2.215386072238 -0.082596273247
H -3.321616352183 0.724510560348 -1.795021001324
H -3.178545601317 -1.812896592891 -1.067193002622
H -1.127841980575 2.056678469874 -1.202081417709
H 1.801156299585 -1.337644426657 0.948074764873
H 3.994412560307 -0.004534330069 1.540456689580
H 3.850474249127 2.532590091349 0.812282191911
Summary of Geometry Optimization
==================================
Opt.Step Energy (a.u.) Energy Change (a.u.) Displacement (RMS, Max)
-------------------------------------------------------------------------------------
0 -1104.216612174516 0.000000000000 0.000e+00 0.000e+00
1 -1104.237771918198 -0.021159743682 9.386e-02 1.493e-01
2 -1104.243530217837 -0.005758299639 7.203e-02 1.274e-01
3 -1104.243363632533 0.000166585304 5.168e-02 9.205e-02
4 -1104.245585316760 -0.002221684227 2.551e-02 4.005e-02
5 -1104.246301651732 -0.000716334971 3.012e-02 4.825e-02
6 -1104.246214246383 0.000087405348 7.350e-03 1.311e-02
7 -1104.246322600028 -0.000108353645 3.811e-03 6.933e-03
8 -1104.246332992981 -0.000010392953 2.786e-03 4.791e-03
9 -1104.246335280611 -0.000002287631 3.040e-03 4.712e-03
10 -1104.246336652051 -0.000001371440 4.744e-04 7.317e-04
11 -1104.246336830749 -0.000000178697 3.190e-04 6.852e-04
Statistical Deviation between
Optimized Geometry and Initial Geometry
=========================================
Internal Coord. RMS deviation Max. deviation
-----------------------------------------------------------
Bonds 0.050 Angstrom 0.114 Angstrom
Angles 3.626 degree 6.170 degree
Dihedrals 0.002 degree 0.005 degree
*** Time spent in Optimization Driver: 764.39 sec
molecule_opt = vlx.Molecule.read_xyz_string(opt_results['final_geometry'])
molecule_opt.show(atom_indices=True)Text file
To optimize an excited state, you need to use the task optimize and to specify which state you want to optimize with the state_deriv_index: keyword in the @gradient section.
@jobs
task: optimize
@end
@response
property: absorption
nstates: 2
@end
@gradient
state_deriv_index: 1
@end
@method settings
xcfun: b3lyp
basis: def2-svp
@end
@molecule
charge: 0
multiplicity: 1
xyz:
...
@end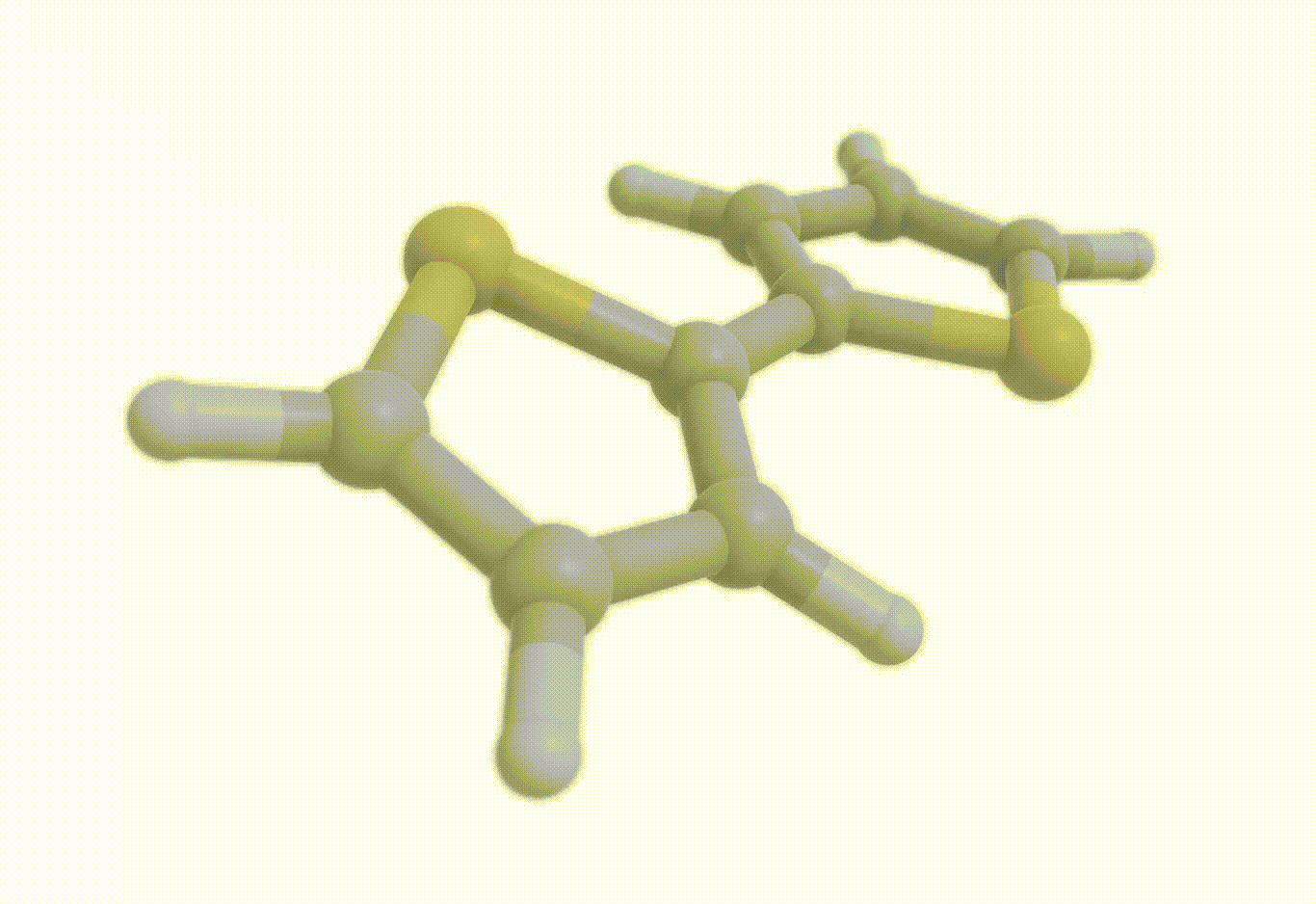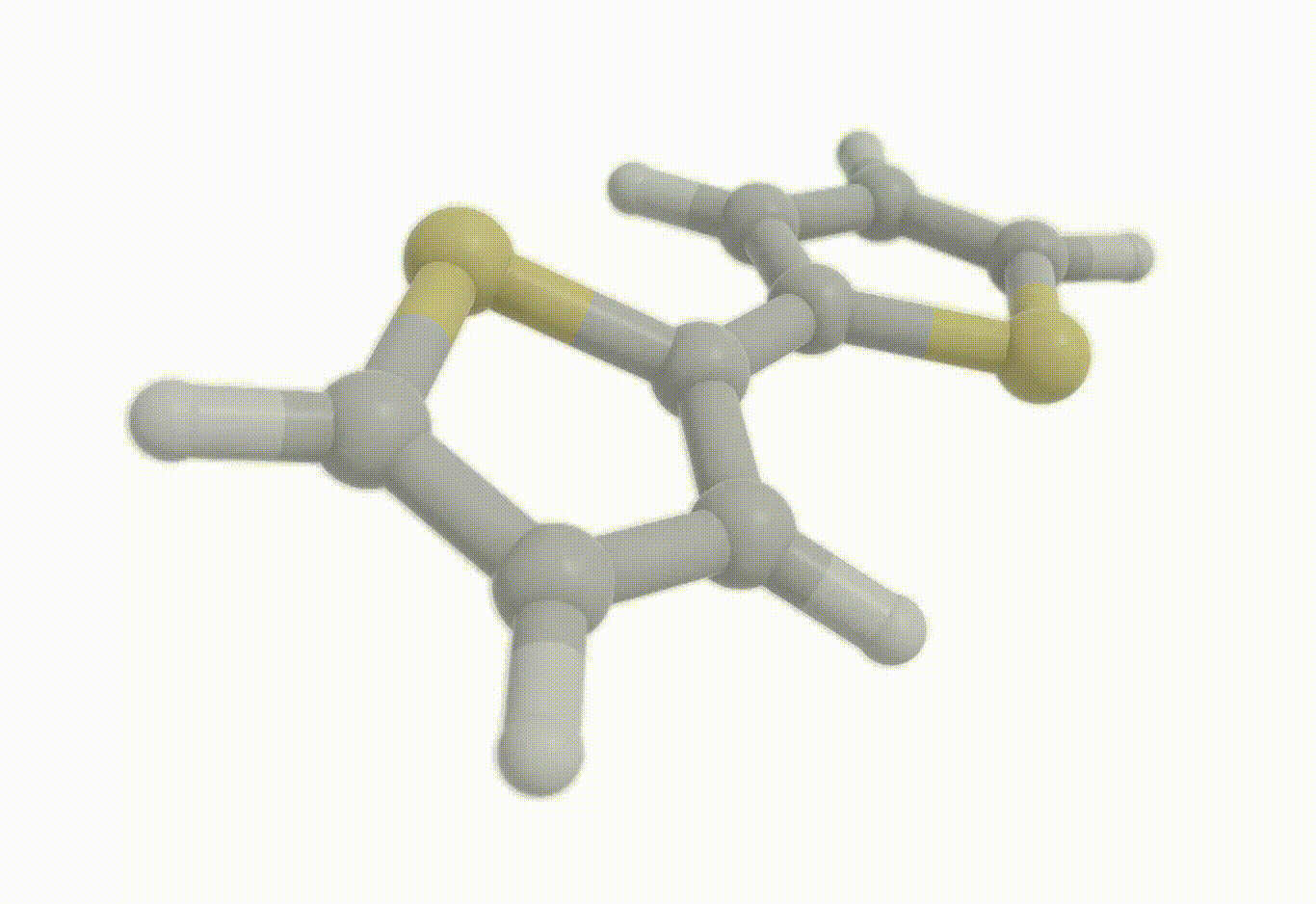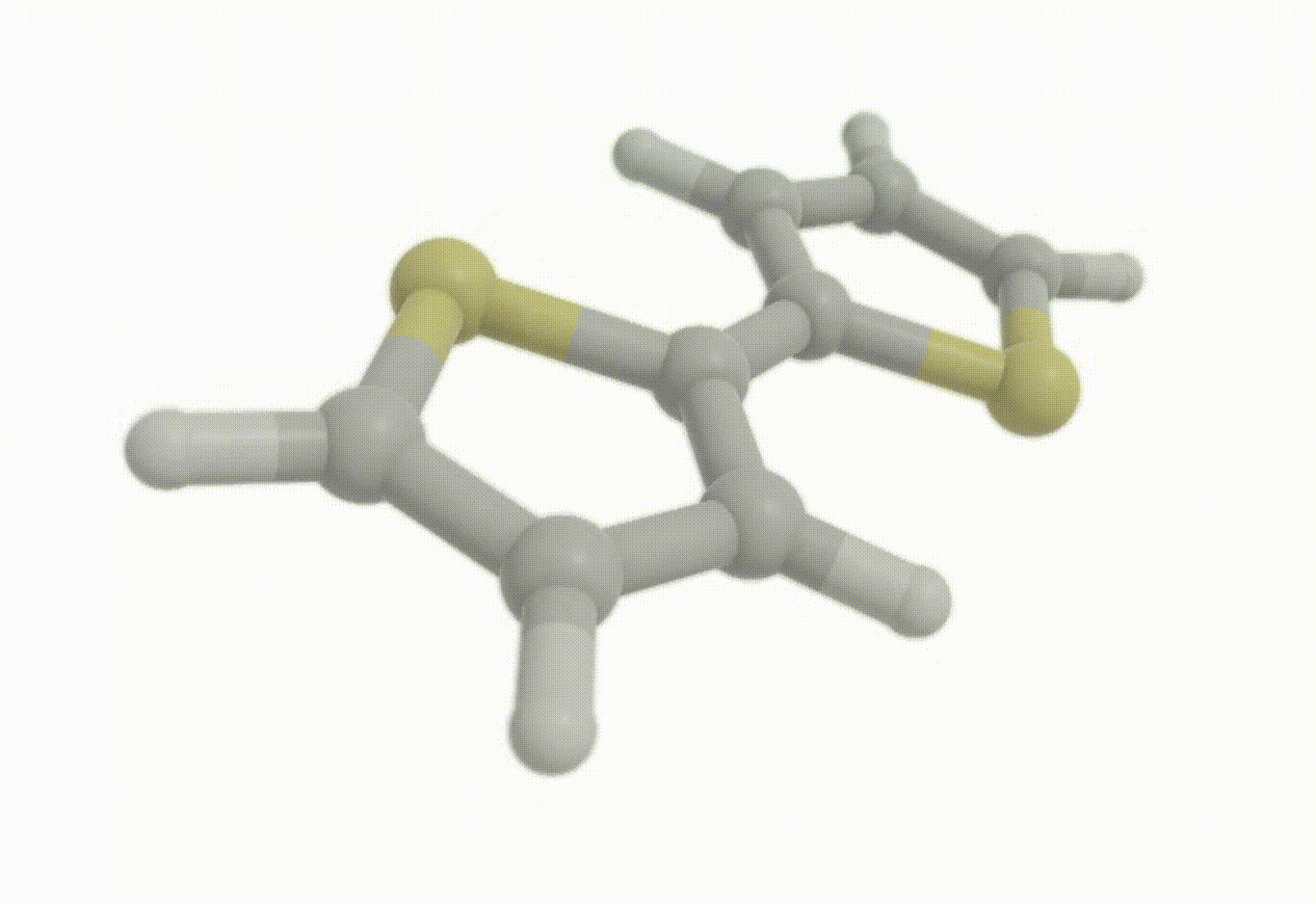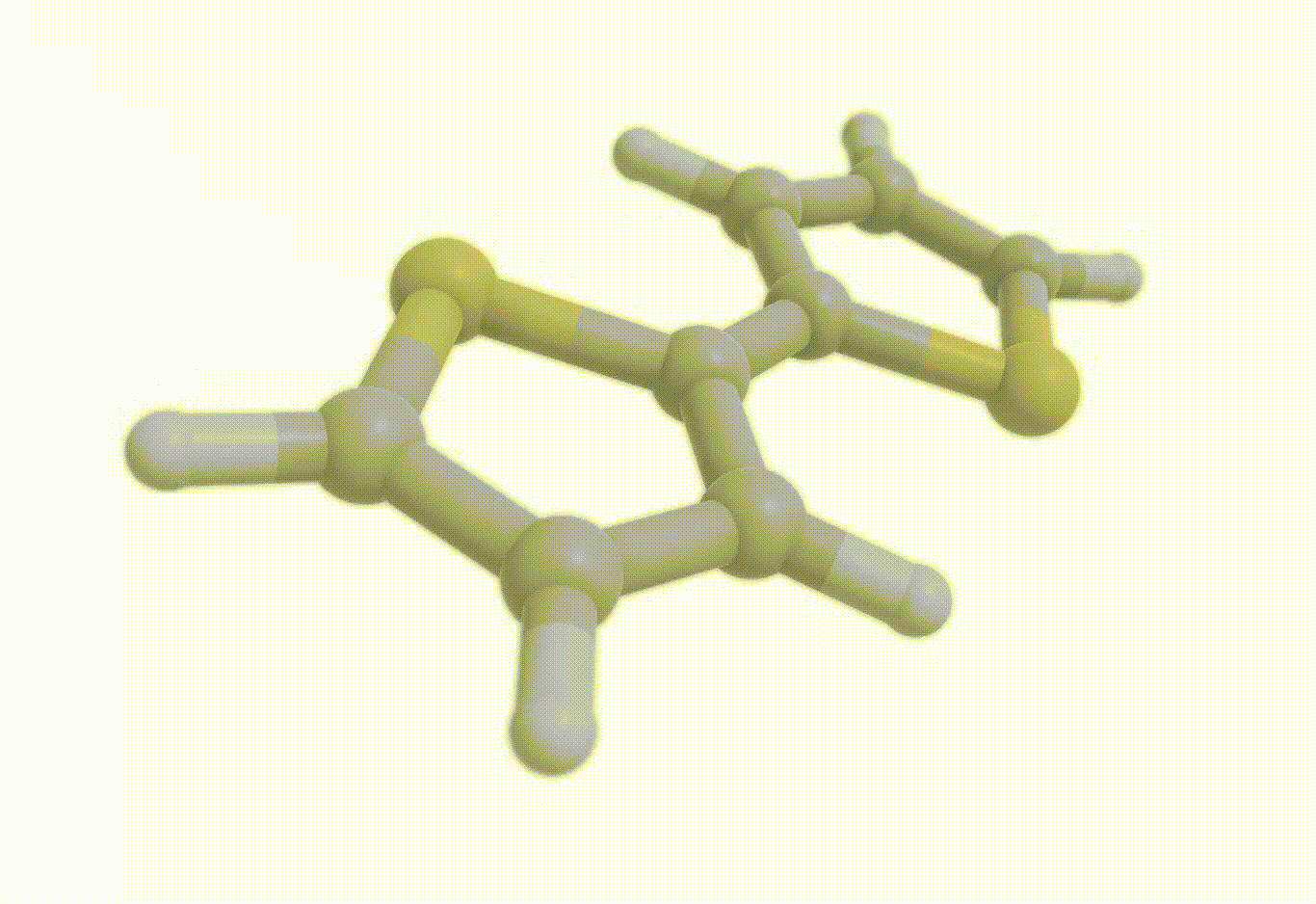
Figure: Optimization of the molecular structure in the excited state, S1.